Bleeding Disorders 101
Congenital bleeding disorders are characterised by impaired blood clotting, usually due to an inherited deficiency in or abnormal functioning of a clotting factor. There are many types (depending on which clotting factor is affected) but, with three exceptions, they are rare.
The major bleeding disorders
The most frequent bleeding disorders are haemophilia A, haemophilia B and von Willebrand disease [Srivastava et al, 2013; Laffan et al, 2011]
Haemophilia A
- Frequency about 1 in 10,000 births
- Due to a deficiency of Factor VIII caused by mutation(s) in the gene F8
- Accounts for 80 – 85% of cases of haemophilia
Haemophilia B
- Frequency is about 1 in 30,000 – 50,000 births [Srivastava et al, 2013; Laffan et al, 2011]
- Due to a deficiency of Factor IX caused by mutation(s) in the gene F9
- Accounts for 15 – 20% of cases of haemophilia
- Traditionally believed to cause a less severe bleeding disorder than haemophilia A but recent evidence suggests there may be no difference among children (Clausen et al, 2014)
Most people with haemophilia are male but it can affect a girl whose father has the disorder and whose mother is a carrier. Females who are carriers can have levels of FVIII or FIX that are 50% or more below normal. The incidence of haemophilia is independent of ethnicity and geography.
Acquired haemophilia has a frequency of about 1.5 per million population. It is caused by autoantibodies that neutralise FVIII cofactor activity and is associated with older age, pregnancy and autoimmune disorders.
von Willebrand disease (VWD)
von Willebrand disease may cause a very mild bleeding disorder that in most cases is undiagnosed, therefore the prevalence is uncertain (but may lie in the range 0.6 – 1.3%) [Laffan et al, 2014; Leebeek and Eikenboom, 2016].
- Caused by a deficiency in, or impaired functioning of, von Willebrand factor (VWF)
- Levels of FVIII activity may also be reduced
- There are three types:
- Type 1 accounts for 70 – 80% of cases and usually causes very mild symptoms
- Type 2 is caused by defective functioning of VWF and is a moderate bleeding disorder; there are four subtypes (2A, 2B, 2M, 2N) depending on how the function of VWF function is altered and which mutation is involved
- Type 3 is due to a near complete or complete absence of VWF and is a severe bleeding disorder
Rare bleeding disorders
The rare bleeding disorders include [Bolton-Maggs et al, 2004]:
- Inherited abnormalities of fibrinogen
- Prothrombin deficiency
- Factor V deficiency
- Combined deficiency of factors V and FVIII
- Factor VII deficiency
- Factor X deficiency
- Inherited deficiency of the vitamin-K dependent clotting factors
- Factor XI deficiency
- Factor XIII deficiency
- Ehlers-Danlos syndrome.
Complications of bleeding disorders
For those with bleeding disorders, prolonged bleeding occurs after surgery or trauma and is normally readily apparent. However, bleeding may also occur covertly and without apparent cause (‘spontaneously’). Bleeding into joints causes long term damage and pain.
In people with haemophilia:
- bleeding most often occurs into joints (70 – 80% of episodes)
- bleeding into muscle accounts for 10 – 20% of episodes
- intracranial bleeding makes up fewer than 5% of episodes
- other sites of bleeding include mucous membranes in the mouth, gums, nose (epistaxis) and genitourinary tract (menorrhagia), the gastrointestinal system (including mouth and throat)
- the pattern of bleeding episodes differs between VWD and haemophilia (type 3 VWD may be more frequently associated with mucosal bleeding than moderate haemophilia; joint and muscle bleeding may be less frequent with VWD) but, with similar bleeding severities, the frequency of joint function loss is comparable.
The severity of bleeding episodes and the risk of spontaneous bleeding depend on the extent to which levels of clotting factor are reduced (or their function is impaired).
| Bleeding severity in haemophilia | Level of clotting factor
(% of normal) |
Bleeding episodes |
| Severe | <1%
(<0.01 IU/ml) |
Spontaneous bleeding into joints or muscles, predominantly without obvious cause. |
| Moderate | 1 – 5%
(0.01–0.05 IU/ml0 |
Occasional spontaneous bleeding; prolonged bleeding with minor trauma or surgery. |
| Mild | 5 – <40%
(0.05–0.40 IU/ml) |
Severe bleeding with major trauma or surgery. Spontaneous bleeding is rare. |
Joint haemorrhage (haemarthrosis)
Bleeding may occur into one or more joints. The presence of iron in the joint adversely affects the maintenance of the synovium, causes loss of cartilage and provokes inflammation [Acharya, 2012]. With continued exposure, the joint becomes painful and loses movement; eventually, bone mineral density is lost. Haemarthroses affect the ankle, knee and elbow more often than the shoulder, wrist or hip (Srivastava et al, 2013].
In the child with severe hemophilia, the first haemarthrosis typically occurs when the child begins to crawl and walk: usually before 2 years of age, but occasionally later. If inadequately treated, repeated bleeding will lead to progressive deterioration of the joints and muscles, severe loss of function due to loss of motion, muscle atrophy, pain, joint deformity, and contractures within the first one to two decades of life.
Clinically, bleeding into the joint causes local warmth or other abnormal sensation (which may be described as an ‘aura’), tenderness and discomfort on movement. This progresses to swelling, pain at rest and extreme loss of movement. In recognition of the risk of long-term complications, a joint affected by three or more spontaneous bleeds within a 6-month period is labelled a target joint [Srivastava, et al, 2103]
In severe haemophilia, the occurrence of joint bleeding increases with age and accounts for:
- 21% of all haemorrhages at 1–6 years
- 50% at 10–17 years
- 60% at 18–65 years [Peyvandi et al, 2016].
A quarter of people with moderate haemophilia have joint bleeds similar to severe haemophilia and about the same proportion appear not to experience joint bleeds [Peyvandi et al, 2016].
The frequency of joint bleeds was greater historically than is currently the case because earlier and more widespread use of prophylaxis has reduced bleeding episodes. A 2001 survey of people with moderate haemophilia found that 43% reported joint impairment and 24% needed orthopaedic aids but bleeding was much less frequent among individuals born after 1970 [den Uijl, et al, 2009].
Almost a quarter of people with VWD surveyed in the early 2000s said they had joint bleeds [van Galen et al, 2015], but over half of those with type 3 VWD were affected [Leebeek et al, 2016].
The table summarises management options for joint damage secondary to bleeding [Srivastava et al, 2013].
| Synovitis | Protect with a removal splint or compressive bandaging
Restrict activities until swelling and temperature of the joint normalise Acute treatment with a COX-2 inhibitor may be useful to reduce pain and swelling The goal of treatment is to deactivate the synovium as quickly as possible and preserve joint function Factor concentrate replacement, ideally given with the frequency and at dose levels sufficient to prevent recurrent bleeding Physiotherapy COX-2 inhibitor may be used to relieve pain Synovectomy should be considered if chronic synovitis persists with frequent recurrent bleeding not controlled by other means |
| Arthropathy | Goals of treatment are to improve joint function, relieve pain, and assist the patient to continue/resume normal activities of daily living
Treatment options depend on stage of arthropathy and the patient’s condition, quality of life and functional limitation COX-2 inhibitor may be used to relieve arthritic pain Supervised physiotherapy aiming to preserve muscle strength and functional ability is very important; secondary prophylaxis may be needed if this causes recurrent bleeding Casts, aids, braces and home adaptations may be needed Surgery and rehabilitation |
| Pseudotumours | Evaluate with MRI and CT scan
6-week course of treatment with clotting factor then repeat MRI; if the tumour is decreasing, continue with factor and repeat MRI for three cycles Aspiration of the pseudotumour followed by injections of fibrin glue, arterial embolisation, or radiotherapy may heal some lesions; surgery may be needed for others |
| Fracture | Immediate factor concentrate replacement, raising levels to at least 50% and maintaining for 3 – 5 days
Use splints, not circumferential plaster Prolonged immobilisation can lead to significant limitation of range of movement in the adjacent joints and should be avoided Physiotherapy should be started as soon as the fracture is stabilised to restore range of motion, muscle strength, and function |
Muscle haemorrhage
This usually occurs after trauma or a sudden stretch, causing blood to leak into a muscle; this may lead to significant blood loss [Srivastava et al, 2013; Rodriguez-Merchan 2008]. The immediate signs and symptoms are ache, pain on movement (stretching or contracting), tenderness on palpation and swelling; these may be less obvious when a deep muscle is affected. Accumulation of blood within the muscle compartment can cause a marked increase in pressure (compartment syndrome) that may be limb-threatening; this requires prompt decompression. Complications include contracture, re-bleeding, pseudotumour and permanent loss of function. There is no published evidence on the incidence of muscle bleeding in people with haemophilia or VWD.
Mucocutaneous bleeding
Bruising, epistaxis, menorrhagia and bleeding from superficial wounds (e.g. after immunisation) are frequently the first signs of a bleeding disorder among children and young people with VWD [Sanders et al, 2015] and are common in carriers of haemophilia A [Paroskie et al, 2015]. Reliable statistics are lacking on the frequency of these relatively minor episodes among individuals with moderate or severe bleeding disorders.
Head injury/intracranial bleeding
All head injuries should be managed as if intracranial bleeding has occurred, requiring an immediate increase in factor level after trauma or when early signs develop. This is adjusted in light of investigations; a confirmed bleed requires maintenance of levels for 10 – 14 days and consideration of secondary prophylaxis for 3 – 6 months.
Viral and bacterial infection
Haemophilia per sedoes not increase the risk of microbial infection and, following the experiences of the 1990s, the risk of transmitting human immunodeficiency virus (HIV), hepatitis C (HCV) and hepatitis B (HBV) through blood products to treat haemophilia has now been virtually eliminated (though risks remain for non-lipid enveloped viruses and prions). When virus-inactivated blood products are not available, every patient should be tested for HIV and HBV at least every 6 – 12 months and whenever clinically indicated. Subsequent management – diagnosis, counselling, treatment and monitoring – is the same as in people who do not have haemophilia. There are no contraindications to HIV therapies in people with haemophilia.
Individuals who do not have HBV immunity should be offered vaccination against HBV and seroconversion confirmed (if seroconversion does not occur, repeat with a double dose of vaccine). Active HBV infection is treated as for people who do not have haemophilia.
The introduction of a new generation of oral direct-acting antivirals (DAAs) has revolutionised the treatment of HCV infection in recent years, offering exceptionally high cure rates and freedom from treatment with interferon [Seifert et al, 2015]. The WFH guidelines recommend standard treatment for HCV, noting that coinfection of HCV genotype 1 and HIV predict a poorer response to treatment; when HCV cannot be eradicated, the patient should be monitored every 6 – 12 months for end stage liver disease. This advice predates the introduction of DAAs and may not represent what can be achieved clinically today. The DAAs are relatively expensive but have been recommended by NICE and the Scottish Medicines Consortium for use in the NHS.
People with haemophilia experience a high frequency of venous access and surgical interventions, creating opportunities for bacterial infection. A clean environment for home therapy is especially important.
Inhibitors
Inhibitors are now considered the most severe treatment-related complication of haemophilia [Srivastava et al, 2013].
- An inhibitor is a high affinity IgG antibody that specifically neutralises the procoagulant activity of a clotting factor.
Inhibitors make it difficult to manage bleeding. In people with haemophilia, they are associated with increased morbidity and disability and they increase the risk of death by a factor of 5 in people with non-severe haemophilia A and by 70% in those with a severe disorder [Peyvandi et al, 2016]. Perhaps contrary to expectations, recombinant factors are associated with a 2-fold higher risk of inhibitor development compared with factors derived from blood [Peyvandi et al, 2016; Collins et al, 2014].
Inhibitors develop in 5 – 10% of people with type 3 VWD who receive multiple transfusions of clotting factor [Laffan et al, 2014].
In haemophilia, risk factors for developing an inhibitor are related to the patient and the treatment [Peyvandi et al, 2016].
| Patient-related factors
|
Treatment-related factors
|
| Severity of haemophilia
F8 gene mutation Family history of inhibitors Being of black ethnic origin Polymorphisms of immune-response genes |
Number of exposure days
Intensity of exposure Product type—plasma derived versus recombinant Age at first exposure Prophylaxis with factor replacement therapy |
The clinical impact of inhibitors depends on the blood level of the inhibitor (titre) and the patient’s anamnestic response:
- Some patients do not readily develop increased levels of inhibitors when retreated with a clotting factor (‘low responders’); in such cases, the inhibitor can be overcome by a higher dose of clotting factor
- a low responder is defined as an inhibitor level persistently <5 Bethesda Units/ml (BU/ml)
- By contrast, ‘high responders’ rapidly develop increased levels of inhibitors on re-exposure
- a high responder is defined as an inhibitor level of ≥5 BU/ml
- high responders require a bypassing agent to nullify the inhibitor or immune tolerance induction to eradicate it
The pattern of inhibitor development is different according to haemophilia severity.
In severe haemophilia:
- about 30% of patients with severe haemophilia A develop FVIII, usually during the first 20–30 days of exposure.
- in severe haemophilia A, the median age of inhibitor development is ≤3 years
- in severe haemophilia B, inhibitors appear early (after 9 – 11 days); the cumulative incidence of inhibitors is 4 – 5% but >80% of those affected are high responders. The development of inhibitors to FIX is associated with anaphylaxis in about half of patients
In non-severe haemophilia:
- the incidence of inhibitors is 5 – 10%
- in non-severe haemophilia A, the median age of inhibitor development is about 30 years, often following intensive FVIII exposure associated with surgery
- the inhibitor may neutralise the patient’s endogenous FVIII, converting their haemophilia from mild/moderate to severe (with a similar pattern of bleeding)
Screening for inhibitors
Because inhibitors increase the risk of bleeding, all patients treated with a clotting factor should be screened. Children should be screened more frequently than adults:
- children – screen every 5 exposure days up to 20 exposure days, then every 10 exposure days for exposure days 21 – 50, then at least twice yearly until 150 exposure days
- adults with >150 exposure days – any failure to respond to adequate factor concentrate replacement therapy in a previously responsive patient is an indication to assess for an inhibitor
- all patients who have been intensively treated for more than 5 days should be screened within 4 weeks of the last infusion
- all patients – prior to surgery, if recovery assays are not as expected and when clinical response to treatment of postoperative bleeding is sub-optimal
Overcoming inhibitors
Inhibitors pose a threat to the management of events such as bleeding into joints. Patients who are low responders may be treated with a much higher dose of the clotting factor. In an emergency, individuals with a low titre of a high responding inhibitor can successfully be treated until the anamnestic response occurs (3 – 5 days).
There are two strategies for treating bleeding in the presence of high titres of high-responding inhibitors in patients with haemophilia: bypassing agents and immune tolerance induction.
- A bypassing agent circumvents the need for factors VIII and IX by generating thrombin through another mechanism
- Two equally effective options are available: single-dose activated prothrombin complex concentrate (APCC) and two doses of recombinant activated FVII
- Some patients respond better to one option than the other; APCC induces an anamnestic response in about 30% of patients with FVIII inhibitors
Immune tolerance can be induced by frequent exposure to a clotting factor. This involves administration of 50 – 200 IU/kg every 1 – 2 days for several months, achieving eradication of inhibitors in 60 – 70% of patients with severe haemophilia A [Peyvandi et al, 2016]. Success has been limited in patients with mild or moderate haemophilia A and in haemophilia B, especially inpatients at risk of anaphylaxis.
A further alternative is to switch to a different FVIII product, which may not be affected by inhibitors to the predecessor. Patients should be monitored for inhibitor development after switching.
Options for patients with VWD who develop an inhibitor include very high doses of recombinant FVIII, recombinant activated FVII, platelet transfusion and tranexamic acid.
Genetic basis of inheritance of the major bleeding disorders
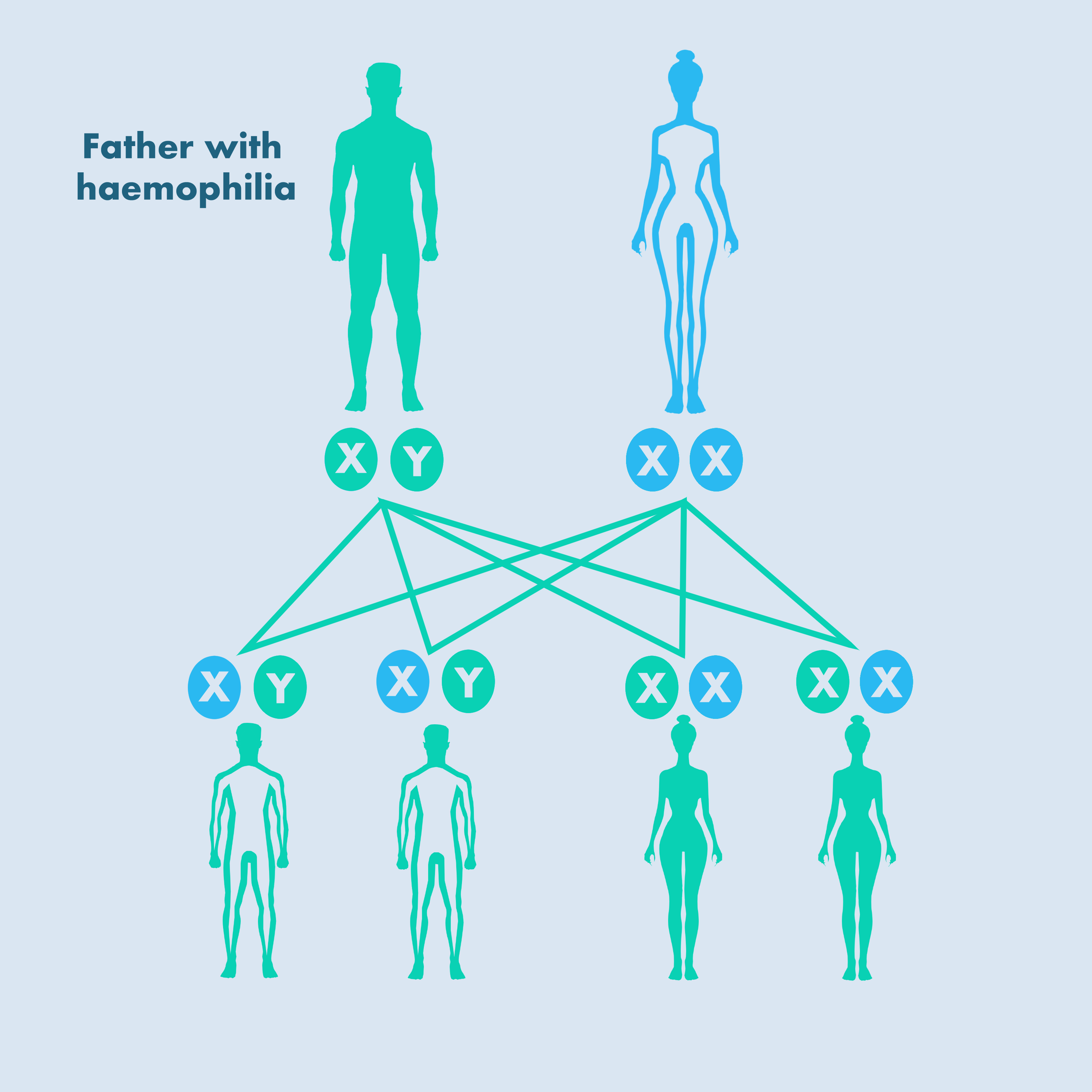
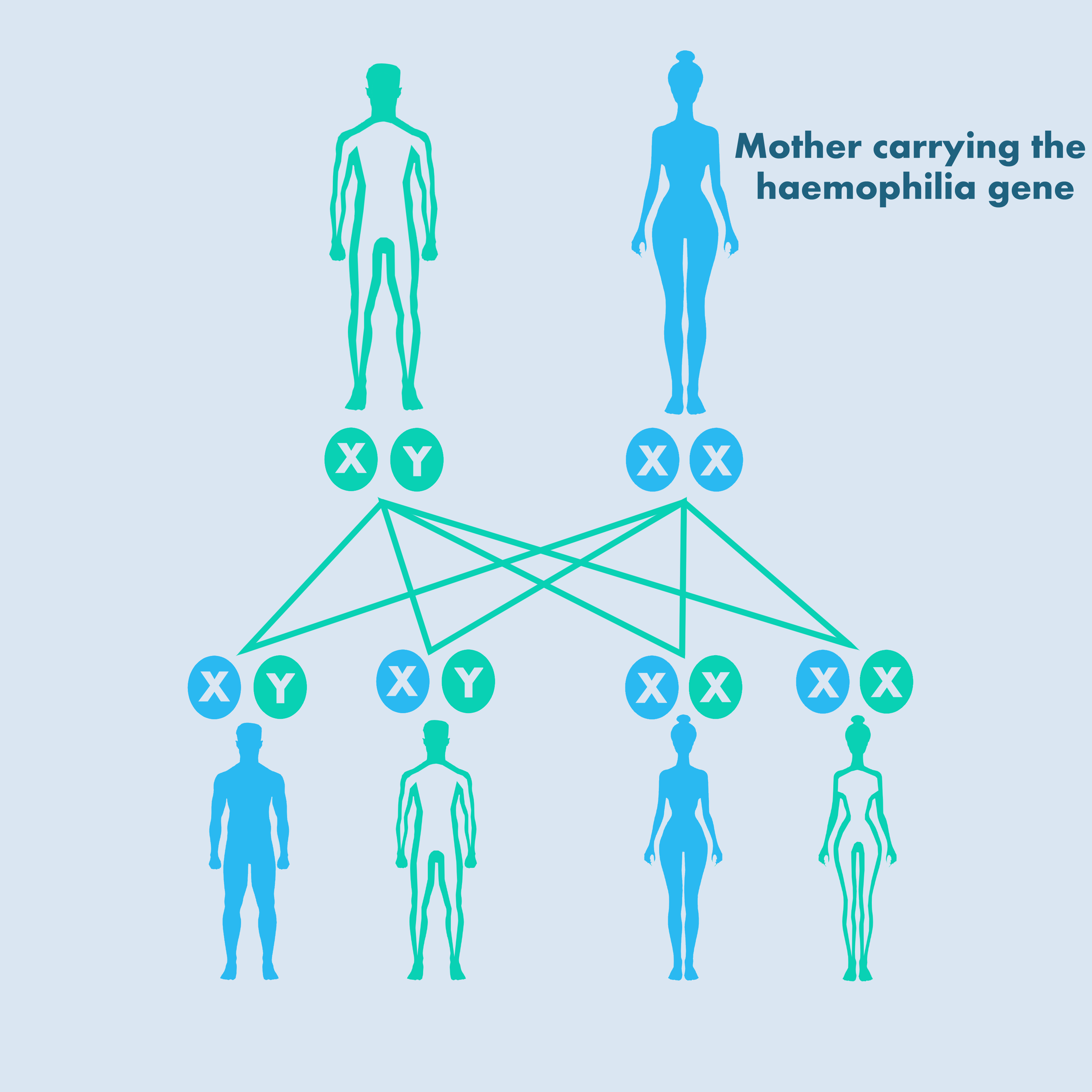
Both haemophilia A and B are X-linked recessive disorders – that is, the gene mutation occurs only on the X chromosome [WFH, 2016]:
- A father with haemophilia does not pass the disorder on to his sons but all his daughters become carriers.
- If a mother carries the mutation, her son has a 50% chance of having haemophilia and her daughter has a 50% chance of being a carrier.
- Up to one-third of cases are due to new gene mutations and there is no family history of haemophilia.
Von Willebrand disease has an autosomal inheritance pattern, meaning the affected gene is not on a sex-linked gene and men and women are equally affected. However, more women than men are diagnosed because they experience more clinical bleeding events (e.g. menorrhagia) [Leebeek and Eikenboom, 2016].
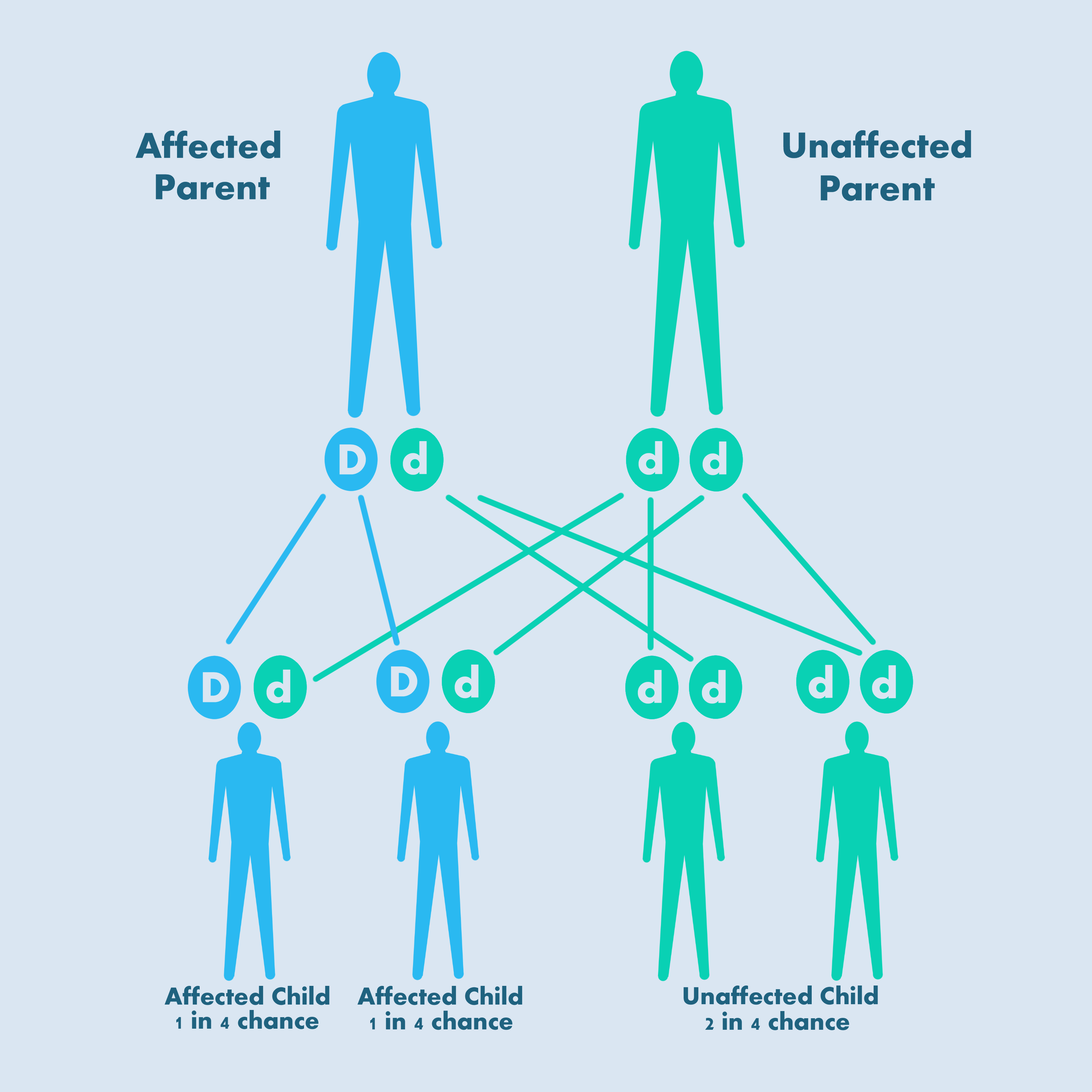
Type 1 VWD and subtypes 2A, 2B and 2M have a dominant inheritance pattern: a child who inherits a normal gene from one parent (‘d’ in the figure) and an abnormal gene (‘D’ in the figure) from the other will develop VWD.
Dominant autosomal inheritance
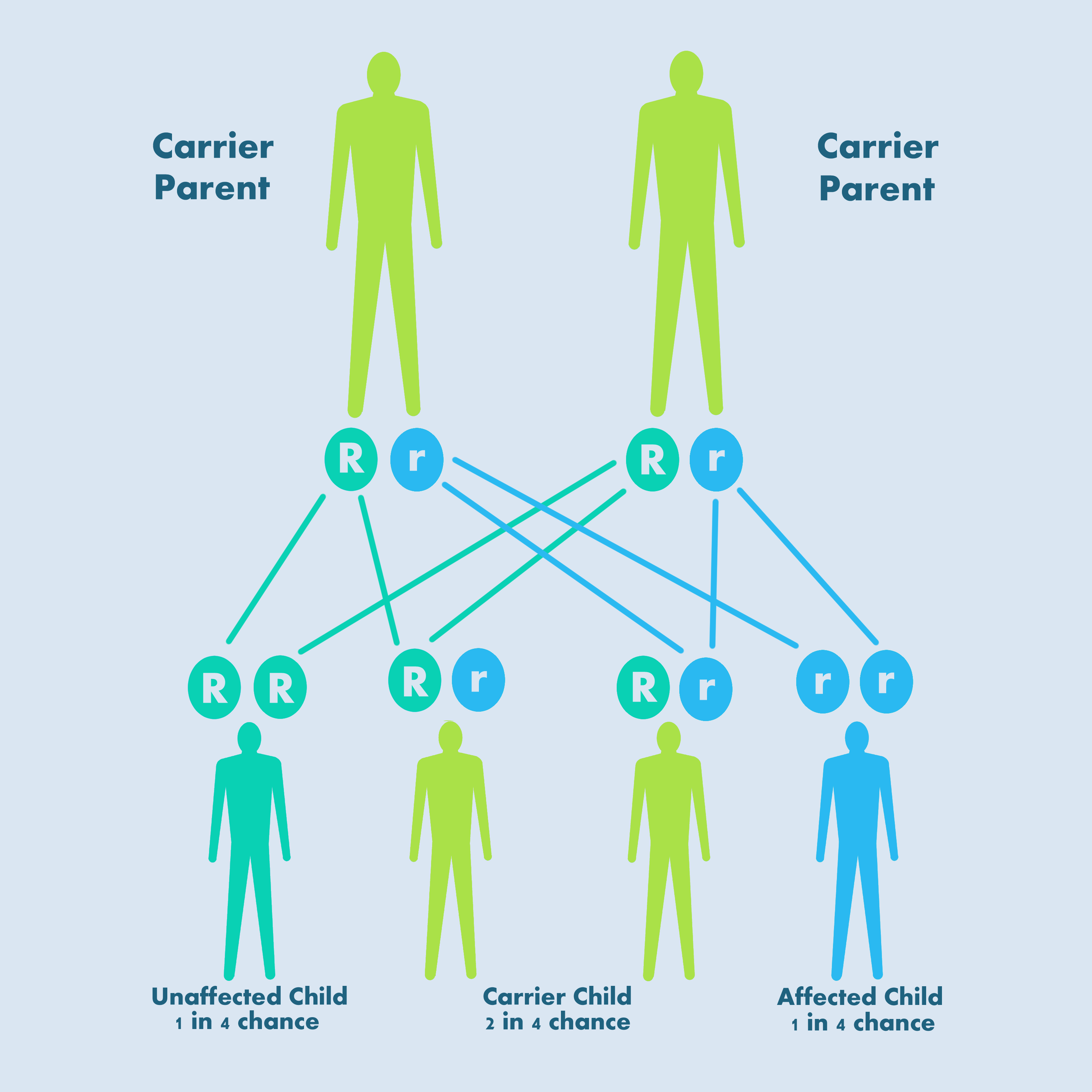
Subtype 2N and type 3 VWD have an autosomal recessive inheritance pattern, meaning a child has to inherit an abnormal gene (‘r’ in the figure) from both parents to develop VWD; if they inherit one abnormal gene, they are a carrier.
Recessive autosomal inheritance
Laboratory diagnosis
The bleeding tendency in mild haemophilia may not be known until the person undergoes surgery or is subject to trauma but even a severe bleeding disorder may not be recognised in a child until they begin to walk.
The diagnosis of a bleeding disorder requires clinical assessment supported by accurate laboratory measures. This has two implications:
- The laboratory should following strict protocols and procedures requiring knowledge and expertise in coagulation testing, use of the correct equipment and reagents and adherence to internal and external quality assurance procedures
- Clinicians should provide samples obtained according to protocol and using the specified equipment and disposables; this includes preparing the patient (e.g. discontinuing aspirin, avoiding strenuous exercise prior to sampling)
Clinicians should adhere to local procedures for venesampling. Standards for collection and processing of biological samples are provided by the WFH.
Typically, a patient will be screened using routine tests before having an assay to determine clotting factor activity. The interpretation of these tests is summarised in the table.
| Possible diagnosis | Prothrombin time | Activated partial thromboplastin time* | Bleeding time | Platelet count |
| Haemophilia A or B** | Normal | Prolonged* | normal | Normal |
| VWD | Normal | Normal or prolonged* | Normal or prolonged | Normal or reduced |
| Platelet defect | normal | normal | Normal or prolonged | Normal or reduced |
* Results are highly dependent on the laboratory method used; ** The same pattern can occur in the presence of FXI, FXII, prekallikrein or high molecular weight kininogen deficiencies
A clotting factor assay will be carried out to determine the diagnosis and to monitor treatment. Tests using pooled normal plasma (PNP) are used to determine whether prolonged coagulation times are due to factor deficiency or circulating anticoagulants of inhibitors. A prolonged APTT that is not fully corrected by mixing the patient’s plasma with PNP suggests the presence of an inhibitor.Confirmation requires a specific inhibitor assay.