Open navigation
Landmarks in haemophilia care
"The more you know about the past, the better you are prepared for the future”
Pre-1400

200AD
First written reference to a bleeding tendency read more
The Babylonian Talmud, written in the 5thcentury and recording a ruling made in the 2ndcentury, is acknowledged as the source of the first documented mention of a bleeding disorder that may have been haemophilia.
The reference comes in the form of advice in the Yevamot tractate,1in a section that primarily addresses inconsistency in rabbinical statements – specifically, whether an event should occur two or three times before being accepted as proof. Rabbi Judah the Patriarch stated:
If a woman circumcised her first son and he died as a result of the circumcision, and she circumcised her second son and he also died, she should not circumcise her third son, as the deaths of the first two produce a presumption that this woman’s sons die as a result of circumcision.
However, Rabbi Simon ben Gamaliel affirmed a general point of Talmudic law: that three events are required:
She should circumcise her third son, as there is not considered to be a legal presumption that her sons die from circumcision, but she should not circumcise her fourth son if her first three sons died from circumcision.
In the 11thcentury, Rabbi Isaac Alfasi confirmed Rabbi Judah’s view.2
The 12thcentury physician Moses Maimonides, in a commentary again focusing on the risks of circumcision, noted that children may be at risk through their mother, even when they have different fathers; transmission of the risk (not haemophilia) via the father was recognised in the 16thcentury by Rabbi Joseph Karo.2
The first description of haemophilia is credited to the surgeon Al-Zahrawi (936‒1013 AD), also known as Albucasis.3-5He described a fatal ‘blood disease’ affecting only males, in which minor trauma provoked prolonged bleeding. Observing the pattern of occurrence in one village, he deduced that the disorder was inherited.
The Book of Matthew in the New Testament of the Bible mentions a woman with a lifelong bleeding disorder who was healed by Jesus. The King James Bible states:6
And, behold, a woman, which was diseased with an issue of blood twelve years, came behind him, and touched the hem of his garment:
For she said within herself, if I may but touch his garment I shall be whole.
But Jesus turned him about, and when he saw her, he said, Daughter, be of good comfort; thy faith hath made thee whole. And the woman was made whole from that hour.
References
- Yevamot 64b. Babylonian Talmud. William Davidson Edition (accessed 26 February 2019).
- Rosner F. Hemophilia in the Talmud and rabbinic writings. Ann Intern Med 1969; 70: 833-7.
- Kaadan AN, Angrini M. Who discovered haemophilia? (accessed 26 February 2019).
- Rosendaal FR, Smit C, Briët E. Hemophilia treatment in historical perspective: a review of medical and social developments. Ann Hematol 1991; 62: 5-15.
- Ingram GI. The history of haemophilia. J Clin Pathol 1976; 29: 469-79.
- Matthew 9:20-22. King James Bible (accessed 26 February 2019).
1400-1799




1450
Johannes Gutenberg pioneers the modern printing press read more
Control of the distribution of knowledge is a powerful asset, both to the purveyors of knowledge and the people who provide the means to disseminate it. The German craftsman Johannes Gutenberg developed the first printing press in secret – not to avoid suppression by the elite of the age fearful of a threat to their power, but from his business partners who wanted a share of the proceeds.
Gutenberg was an inventor and entrepreneur. According to Encyclopaedia Britannica, he financed his activities using advances from three investors.1They agreed a contract stating that, in the event of a death, his heirs would be compensated financially but would not inherit any shares in the enterprise. When one died, his family challenged the contract in court; they lost but the proceedings revealed Gutenberg had been concealing his work on a printing machine from his investors. His innovations included a soft metal alloy to make reusable type, an oil-based ink and a press that could apply even pressure to vellum or paper.
In 1450, keen to refine his designs, Gutenberg subsequently borrowed more from Johann Fust, a financier in Mainz, Germany. But it seems Gutenberg was a perfectionist and Fust, weary of waiting for a return on his investment, successfully sued Gutenberg for repayment with interest. The court documents have been preserved and provide clear evidence that Gutenberg developed the first known printing press, though he lost it to Fust in 1455.
It is possible that Gutenberg printed indulgences (letters of forgiveness sold by the Catholic Church) as early as 1452. His only major printing enterprise was a Latin edition of the Bible, which he displayed to potential investors in 1454.
The first printed book to bear the title of its printer was a Psalter (a book of psalms for public worship). Published in 1457, it carries the names of Fust and his son-in-law, Peter Schöffer, whom Gutenberg had employed. However, it is believed that Gutenberg may have been involved its production.
Gutenberg died in Mainz in 1468. William Caxton introduced the printing press to London in 1476, predominantly publishing the works of Chaucer, popular poets, chivalric romances and histories, mostly in English.
References
- Lehmann-Haupt HE. Johannes Gutenberg. German printer. Encyclopaedia Britannica. Last updated February 2019 (accessed March 2019).
Further reading
The British Library. Treasures in Full: Gutenberg’s Bible (accessed March 2019).
1747
James Lind conducts the first clinical trial read more
It’s widely known that, in the 19thcentury, the Americans christened British sailors ‘Limeys’ after the Royal Navy’s alleged fondness for stocking the fleet with limes to prevent scurvy during long voyages. The credit for introducing this practice is sometimes laid at the door of James Lind (1716–1794), an Edinburgh physician who served with the Navy between 1738 and 1748. But Lind’s true innovation was to carry out and document one of the earliest prospective controlled clinical trials.
Vitamin C is essential for the production of structurally normal collagen. Deficiency causes skin lesions, bleeding from the mucosae and into joints, anaemia, delayed wound healing, cardiac failure, hypotension and death.1Sailors had known for centuries that consumption of citrus fruit prevented scurvy: the Portuguese explorer Vasco da Gama recorded foraging for citrus fruit in 1498, and it was described by John Woodall, the first surgeon-general of the East India Company, in 1617.2
In May 1747, Lind was serving on the HMS Salisburyas part of a blockade in the English Channel when some of the crew developed scurvy.2He selected twelve and assigned two each to a different treatment popular at that time – cider, elixir vitriol (dilute sulphuric acid), vinegar with meals, sea water, a herbal paste or citrus fruit (two oranges and one lemon, though this continued for only six days before stocks ran out). Lind tried to control for possible variables: the men were as similar as he could find, lived in the same accommodation and ate the same food. Of the two men lucky to have fruit, one was fit for duty after six days and the other was well enough to tend to his comrades.
Lind’s experiment is an example of testing a hypothesis in the scientific way we now recognise as a clinical trial. He described his findings in A treatise of the scurvy. In three parts. Containing an inquiry into the nature, causes and cure, of that disease. Together with a critical and chronological view of what has been published on the subject (1753). The publication also included another innovation: a systematic review of previously published literature.
Despite what seems like convincing evidence, Lind’s finding was not put into practice immediately and the Royal Navy did not order supplies of lemon juice until 1795.3 Surprisingly, Lind never strongly advocated citrus fruit to prevent scurvy and trials of other remedies continued into the late 18thcentury.4 This is an early example of a challenge that innovators face today – translating scientific discoveries into practical interventions.5 Many other factors contributed to ill health aboard ship, including cramped accommodation, poor quality fresh water, and infectious diseases such as malaria, typhus and yellow fever. It may have been difficult to see the efficacy signal amongst all the noise – another problem facing modern science!
References
- Byard RW, Maxwell-Stewart H. Scurvy-characteristic features and forensic issues. Am J Forensic Med Pathol 2019; 40: 43-6. doi: 10.1097/PAF.0000000000000442.
- Milne I. Who was James Lind, and what exactly did he achieve? JLL Bulletin: Commentaries on the history of treatment evaluation. 2012 (accessed 26 February 2019).
- Houlberg K, Wickenden J, Freshwater D. Five centuries of medical contributions from the Royal Navy. Clin Med (Lond) 2019;19:22-5. doi: 10.7861/clinmedicine.19-1-22.
- Lamb J. Captain Cook and the scourge of scurvy. Updated February 2011 (accessed 26 February 2019).
- Rapport F, Clay-Williams R, Churruca K, et al. The struggle of translating science into action: Foundational concepts of implementation science. J Eval Clin Pract 2018; 24: 117-26. doi: 10.1111/jep.12741.
1752
Benjamin Franklin discovers that lightning is electrical read more
It is not readily apparent why the preference for a pointed or blunt lightning rod should be a sign of colonial rebellion ‒ but that, according to the Franklin Institute in Philadelphia, United States, was one result of Benjamin Franklin’s experiments on electricity in 1752.1
Franklin (1706–1790) was a successful publisher who, in 1748, retired from commerce to devote his time to politics and experiments on electricity. He is credited with the discovery that lightning is electricity, but has several other innovations to his name, including the first use of the terms ‘positive’ and ‘negative’ in reference to electrical charges, and the concept of a battery.
Having given himself a substantial shock in the course of his research, Franklin was familiar with the properties of electricity and recognised similarities between the electrical discharges he created in his laboratory and the appearance of lightning. He wasn’t alone in thinking this and, by the 1750s, several scientists had become interested in protecting tall buildings from lightning strikes.
Franklin proposed that a sharply-pointed iron rod could draw a discharge down from a storm and suggested attaching one to the spire of a church that was due to be built (Philadelphia being a largely flat state).2 His plan was presented to the Royal Society in London and greeted with ridicule, but it was successfully carried out in 1751 by the French scientist Thomas-François Dalibard.3
There is some doubt as to whether Franklin actually conducted his experiment himself; however, it is said that, unaware of Dalibard’s success and having lost patience waiting for the church to be built, he resolved that a kite would do just as well. Helped by his son, he attached a pointed rod to the kite and connected it via a metal line to a key in a Leyden jar (a device to store static electricity, similar in principle to a basic capacitor). The men retreated to a dry barn for safety. The kite was not struck by lightning but drew down electrical charge, as Franklin discovered when he moved his hand near the key.
The story goes that Franklin then became a strong advocate of pointed lightning rods, contrary to the prevailing fashion for blunt rods – a fashion supported by no less than the British monarch George III.1 When Philadelphia’s new buildings were equipped with pointed rods, it was perceived as an anticolonial gesture.
Franklin subsequently entered politics. In 1776, he helped to draft and was one of the signatories of the Declaration of Independence and, in 1783, he was a signatory of the Treaty of Paris that ended the American War of Independence.4
References
- Franklin Institute. Franklin’s lightning rod (accessed March 2019).
- Benjamin Franklin Historical Society. Kite experiment (accessed March 2019).
1796
Edward Jenner pioneered the concept of vaccination read more
The World Health Organization (WHO) says that vaccination is arguably the most cost-effective single health intervention we have, saving around 2–3 million lives per year worldwide.1 Edward Jenner (1749–1823) is credited with discovering vaccination but his contribution was (as is the way with scientific innovation) more of an improvement – on current practice albeit a very significant one. It is also a story revealing the lack of humanity shown by the wealthy and educated for people less privileged than themselves.
Smallpox is an infection by the Variola virus that causes fever and a pustular rash, beginning in the mouth and spreading all over the body.2 About 30% of infected people die – historically, reported mortality rates were above 80% among children3‒and those who survive are left with disfiguring scars and sometimes blindness. It is highly infectious. The WHO began a global eradication plan in 1959 and eventually declared the world free of smallpox in 1980.2
Smallpox was introduced into Europe between the 5thand 7thcenturies. It was a major killer, with no respect for class or wealth.3 One of the treatments long practised in Africa, India and China was inoculation – the deliberate introduction of material from a smallpox lesion into the skin of an uninfected person, with the aim of making them immune to infection. Inoculation was introduced into Europe at the turn of the 18th century, where it became known as variolation after the smallpox virus. It was not a benign procedure: 2%‒3% of people subsequently died from smallpox, and others contracted syphilis and tuberculosis from contaminated blood.
Variolation was accepted by English society thanks to the advocacy of Lady Mary Wortley Montague, wife of the British Ambassador to Turkey and herself a survivor of smallpox, who had her son and daughter inoculated by the embassy surgeon Charles Maitland. This sparked interest from the royal family, who permitted an experiment on prisoners in Newgate gaol and on orphaned children. When none died and several did not contract smallpox when exposed to it, Maitland successfully variolated the two daughters of the Prince of Wales in 1722.
Jenner was therefore practising in a culture where variolation was accepted, and he himself had been successfully inoculated at age four. His contribution was to establish the principle of using a safer source of antigenic material. As a country doctor, he had noticed that cow maids did not often develop smallpox. They did, however, catch cowpox from the cattle they milked. Cowpox is a benign infection in humans, causing only a mild fever and an unsightly rash. It was believed that, after recovery from the infection, a person never contracted cowpox again. Jenner tested the hypothesis that infection induced lasting immunity by inoculating eight-year-old James Phipps, his gardener’s son, with cowpox. The boy caught the infection and recovered after a week. Jenner then inoculated him with smallpox. Fortunately for all involved, and for the world generally, James did not develop the infection. Jenner named the new procedure vaccination, after the Latin for cow, vacca.
Jenner reported his experiment to the Royal Society in London to a lukewarm response and demands for more proof. He then vaccinated other children, including his son, and the practice gradually took hold among other practitioners. In 1802, parliament awarded Jenner £10,000 in recognition of his work and a further £20,000 five years later. Variolation was banned in England in 1840 and vaccination with cowpox was made compulsory in 1853.4,5
References
- World Health Organisation. 10 facts on immunization. Updated March 2018 (accessed 26 February 2019).
- Smallpox. Centers for Disease Control and Prevention. Updated July 2017 (accessed 26 February 2019).
- Riedel S. Edward Jenner and the history of smallpox and vaccination. Proc (Bayl Univ Med Cent) 2005; 18: 21–5.
- About Edward Jenner. The Jenner Institute (accessed 26 February 2019).
- Edward Jenner (1749-1823). Brought to Life: Exploring the History of Medicine. Science Museum (accessed 26 February 2019).
1800-1849



1803
Recognition of a haemorrhagic bleeding disorder that affects mostly men read more
The first description of a bleeding disorder that affected only males is usually attributed to John C Otto (1774‒1844), an American physician working in Philadelphia, Pennsylvania.1 However, there seem to have been several reports documenting transmission within a family around that time:
…the anonymous obituarist of Isaac Zoll, writing in 1791 (qu. McKusick 1962), Consbruch in 1793 and 1810, Rave in 1796 (qu. Bullock and Fildes, 1911), and Otto in 1803 all described families in which males suffered abnormally prolonged post-traumatic bleeding. In the Zoll family, six brothers bled to death after minor injuries, but their half-siblings by a different mother were unaffected; in Consbruch’s family, a man and two of his sister’s sons were affected; Rave himself was affected, with his three brothers…2
Otto’s account is a model of scientific caution and precision. He begins by describing a woman named Smith of Plymouth, New Hampshire, who transmitted an ‘idiosyncrasy’ to her descendants, the Smiths and the Shepards: ‘If the least scratch is made on the skin of some of them, as mortal a hemorrhagy will eventually ensue as if the largest wound is inflicted.’ He goes on to describe how persistent bleeding, despite partial healing, rapidly disables the individual and ‘death, from mere debility, then soon closes the scene.’
Otto adds: ‘It is a surprising circumstance that the males only are subject to this strange affection, and that all of them are not liable to it… Although females are exempt, they are still capable of transmitting it to their male children…’
The Smiths were not the only family known to have the condition. Otto cites reports of similar cases described by several of his colleagues and, with great insight, adds: ‘When the cases shall become more numerous, it may perhaps be found that the female sex is not entirely exempt…’
The 1803 report includes a description of the many medicines offered as treatment, including bark, astringents (topically and internally), strong styptics and opiates. Only one worked for Otto’s patients: sulphate of soda (sodium sulphate) ‒, ‘An ordinary purging dose, administered two or three days in succession, generally stops them [haemorrhage]; and, by a more frequent repetition, is certain of producing this effect.’What the mechanism of action could be is a matter for speculation (possibly dehydration and hypotension, secondary to bleeding and purgation). Otto did not understand how this medicine acted but he did not dismiss the family’s experience of treatment, noting that it had worked too often for this to be coincidence and his ignorance was not a good reason to disbelieve them. And, as he pointed out, ‘In every case, however, a doubtful remedy is preferable to leaving the patient to his fate.’
References
- Otto JC. An account of an hemorrhagic disposition existing in certain families. Reprinted from The Medical Repository and Review of American Publications on Medicine, Surgery and the Auxiliary Branches of Science 1803, vol. 6, 1‒4. Clin Orthop Relat Res 1996; 4‒6 (accessed 26 February 2019).
- Ingram GI. The history of haemophilia. J Clin Pathol 1976; 29: 469‒79.
1813
Recognition that men with haemophilia pass the trait to unaffected daughters read
more
The Philadelphia physician John Otto is credited with the first record that haemophilia causes a bleeding disorder only in men. In 1803, he wrote:
‘It is a surprising circumstance that the males only are subject to this strange affection, and that all of them are not liable to it … Although females are exempt, they are still capable of transmitting it to their male children…’1
In the years that followed, accounts were published of several families affected by haemophilia in what was then the 13 British colonies on the east coast of the Americas. Among the most important was a lengthy description of the Appleton-Swain family by John Hay, a physician in Reading, Massachusetts.2 His interest was both professional and personal. Jonathan, his eldest son, married a descendant of Oliver Appleton, the first family member known to be affected, who had lived 100 years previously. Jonathan had eight children, three of whom were sons; at the time of Hay’s report, none had shown signs of exceptional bleeding but he feared that the youngest, at eight years old, ‘has the exact complexion of the bleeders’.
Hay’s description of the fate of the Appletons and Swains makes grim reading. Oliver Appleton died in old age from bleeding from a pressure sore and the urethra. The death of Oliver Swain, one of his grandsons, is recorded in full. Oliver was kicked by a horse, producing a leg wound three inches long and open to the bone. He initially bled profusely, then intermittently rapidly and slowly as physicians tried to stem the flow. He took 19 days to die, in severe pain and ‘in the greatest distress of body’, often lucid but sometimes delirious. Attempts to apply a ligature above the wound failed: ‘the blood has burst out above the ligatures, which caused extreme distress’. Oliver was attended by his brother Thomas, who was a doctor; he later died of a pulmonary haemorrhage. In all, 16 male members of the family were known to have haemophilia, of whom five were known to have died from bleeding.
Though he never explicitly states that Oliver Appleton’s female descendants were carriers, Hay’s painstaking account clearly identifies the women whose sons have haemophilia when they themselves had not apparent bleeding disorder. According to Bulloch and Fildes’ definitive 1911 review of what was known about haemophilia,3 Hay’s report does not provide definitive evidence of haemophilia in all the cases he described (for example, some deaths may not have been related to haemophilia and some family members had no recorded bleeding until adulthood). However, they conclude that his description ‘…bears the stamp of accurate observation. Hay was immediately acquainted with many of the cases and even related to them. Under these circumstances it is highly probable that he forbore to multiply instances.’
References
- Otto JC. An account of an hemorrhagic disposition existing in certain families. The Medical Repository and Review of American Publications on Medicine, Surgery and the Auxiliary Branches of Science 1803;6:1-4 (accessed 29 January 2019)
- Hay J. Account of a Remarkable Hæmorrhagic Disposition, Existing in Many Individuals of the Same Family. New Engl J Med 1813;2:221-5. (Reprinted in The London Medical Physical Journal 1815 Jan;33(191):9-12.
- Bulloch W, Fildes P. Treasury of Human Inheritance. Parts V and VI. Eugenics Laboratory Memoirs XII. University of London Francis Galton Laboratory for National Eugenics. Dulau & Co. London. 1911. (available at https://wellcomelibrary.org).
1828
First use of the term “haemophilia” read
more
It may have been Freidrich Hopf, a German student, who first coined the word haemophiliain his doctoral thesis Über die Hämophilie [About Haemophilia] in 1828. Or perhaps it was his supervisor, Professor Schönlein, because some sources say that Hopf’s new word had actually been haemorrhaphilie and it was deemed too cumbersome. Whichever it was, the journey from conception to widespread use could not have been plain sailing, as other writers at the time suggested many alternatives.
Dr J Whickam Legg, a casualty physician at London’s St Bartholomew’s Hospital, provides a singular account of the acceptance of haemophiliaas the word of choice in his A Treatise on Haemophilia,Sometimes Called the Hereditary Haemorrhagic Diathesis (1872):1
‘In England this disease is often called the haemorrhagic diathesis, or tendency … On the continent, the disease is universally called haemophilia, or haemarrhaphilia. The former of these is the more commonly used; neither is good, but it is now too late to attempt to change.’
Earlier terms had included the German Bluterkrankheit, Blutsuchtand Blutungssucht and the French diathèse hémorrhagique, hémorrhagie constitutionelle and purpura constitionel.
Legg resorts to footnotes for grumpy elaboration:
‘Various other names have been proposed: haemorrhophilia, haematophilia, haemorrhagophilia, idiosyncrasia haemorrhagica and morbus haematicus. A name proposed by Uhde… is amychaemorrhagia… perhaps it is the worst yet suggested.
‘Haemorrhaphilia is the name used by Schönlein in his Vorlesungen (third ed. 1837). Virchow seems to think that Schönlein was the author of the word haemophilia. I am unable to say from my own observation by what writer the name was first used. Die Hämophilie is said to be the title of an inaugural dissertation by Hopf, at Würzburg, in 1828. The word is so barbarous and senseless that it is not wonderful that no one should be proud of it.’
So, with Legg’s grudging acceptance, the term haemophilia – by far the best of the options available ‒ has stuck.
Reference
Legg JW. A Treatise on Haemophilia Sometimes Called the Hereditary Haemorrhagic Diathesis.
London: HK Lewis, 1872 (accessed 26 February 2019).
1840
Blood transfusion used to treat haemophilia read more
In 1839, the eminent German surgeon Johann Dieffenbach developed a new procedure to treat strabismus (squint) that involved dividing the muscles around the eye, so as to reduce their effect on one side and rebalance the tension of opposing muscle groups. It was such a success that, in 1840, the London surgeon Samuel Lane considered it an unremarkable procedure to offer 11 year-old George Firmin. Unfortunately, George was a far from unremarkable child and the complications that followed his operation led to the first recorded use of blood transfusion to treat bleeding due to haemophilia.1
There was more bleeding than usual during surgery, Lane noted, but it subsided and George was able to walk home afterwards. But he soon started bleeding again and the family eventually called the surgeon out that evening, when they belatedly revealed that George had a history of life-threatening bleeding after minor trauma that had resulted in four hospital admissions.
On this occasion, George’s wound continued to bleed for six days and five nights ‘despite the usual remedies, both general and local’. These included applying gum tragacanth mixed with beaver hair to promote coagulation; this was initially successful but the clot formed was weak and soon displaced. By the fifth day, George had lost so much blood that he was unconscious, vomiting and fitting. Lane resolved the boy would not die and he would attempt a blood transfusion at 7pm on the sixth day.
Transfusion was, by contrast with Dieffenbach’s procedure, a rare event at the time due to its poor track record: early attempts at transfusing blood from lambs and calves had, unsurprisingly, proved fatal. Fortunately for George, the obstetrician James Blundell had realised the importance of using human blood and had carried out the first successful transfusion in 1829. He personally explained the procedure to Lane.
Lane describes the procedure in great detail. He collected blood from ‘a stout, healthy young woman’ into a funnel which, via a stop-cock, fed directly into a syringe. Initially the donated blood clotted quickly, a problem solved by excluding air from the syringe chamber. Blood was infused and the syringe rewashed five times in total, after which about half a pint of blood had been donated. Over the next one to two hours, George sat up in bed and drank a glass of wine and water from his own hand. ‘The contrast between life and death could scarcely be greater,’ Lane commented. George recovered fully over the next three weeks (and his strabismus was corrected).
It’s possible that blood transfusion may have been proposed as a treatment for bleeding due to haemophilia as early as 1832 by the German physician Johan Schönlein (who gave his name to Henoch-Schönlein purpura),2but Lane’s is the first definitive account. He and George were fortunate that the donor had compatible blood and that bacterial contamination was somehow averted. Such problems brought a temporary stop to blood transfusion until, at the turn of the century, blood groups were discovered. Subsequent research revealed that blood fractions contained the necessary clotting factors and, in the mid-1930s, the therapeutic potential of plasma precipitate was demonstrated.
References
- Lane S. Haemorrhagic diathesis. Successful transfusion of blood. Lancet 1840; 35: 185-8.
- Schramm W. The history of haemophilia – a short review. Thromb Res 2014; 134 (Suppl) :S4–S9. doi: 10.1016/j.thromres.2013.10.020
1850-1899








1853
Queen Victoria's son, Leopold, is born read more
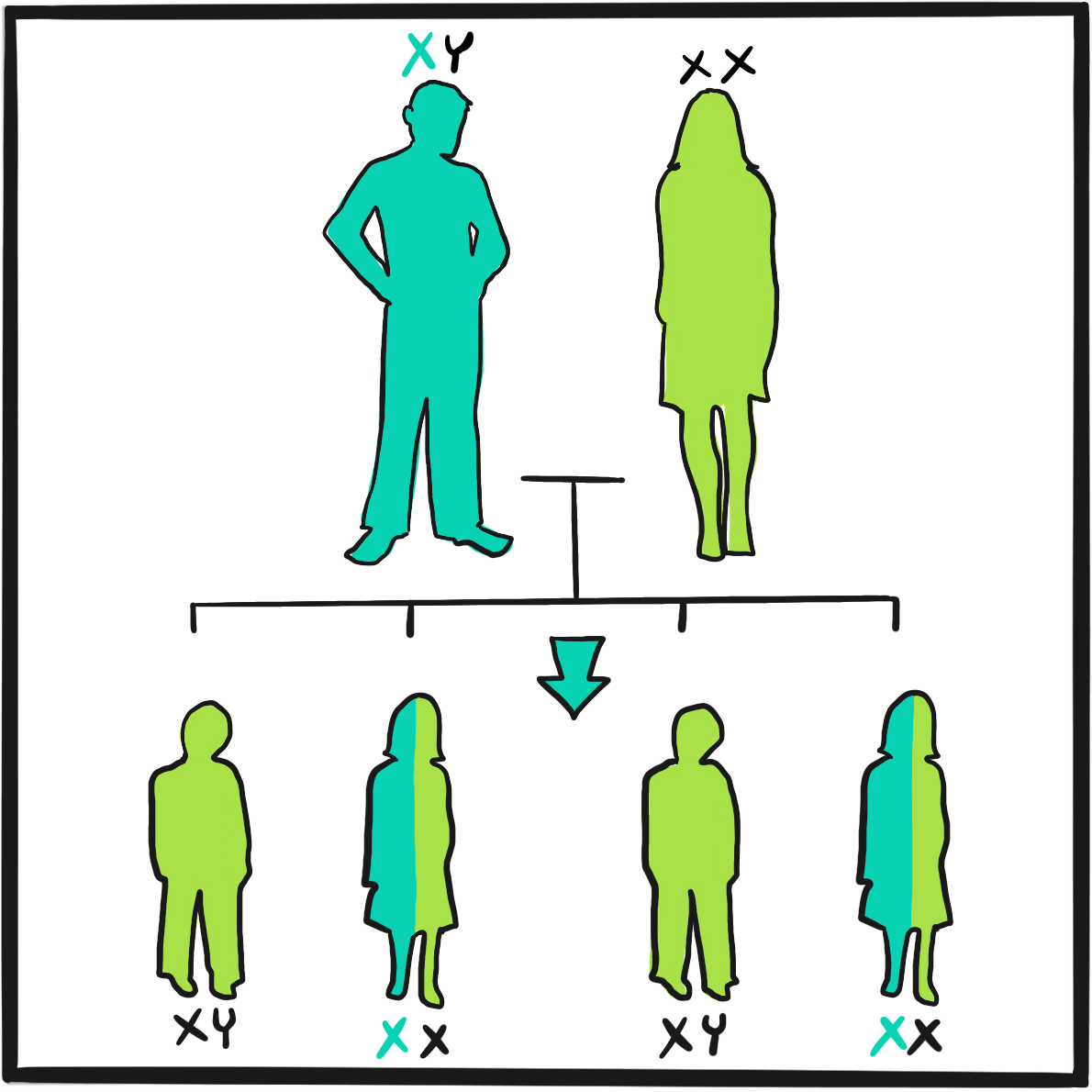
Haemophilia was unusually prevalent among European royalty in the 19thcentury ‒something that is attributed to its emergence in Queen Victoria’s family and the practice of arranging marriages between royal houses to secure power and influence.
Prince Leopold George Duncan Albert, the fourth of Victoria’s nine children, was born on 7 April 1853. He was diagnosed with haemophilia in childhood and was reputedly a delicate child. He died in 1884, probably of a cerebral haemorrhage following a fall. At least two of Victoria’s daughters, Alice and Beatrice, were haemophilia carriers, as was Leopold’s daughter Alice. Their marriages and those of their children resulted in the emergence of haemophilia in the Spanish and Russian royal families. In total, nine or ten of Victoria’s male descendants were known to have had haemophilia; the true number of women who were carriers is not known as some died without having children.
Victorian society did not deal with the issue well. Britain in the mid-19thcentury was, like much of Europe, experiencing revolutionary fervour. In 1848, the Queen was sent to the Isle of Wight to protect her from the Chartist marchers in London. During her reign, she survived six assassination attempts. Haemophilia was seen as a weakness by an establishment that could not afford to yield an inch to republicanism.
In 1868, the medical and lay press carried reports of Leopold’s bleeding episodes. One commentator suggests ‘the disease was an embarrassment to the monarchy, who generally kept as quiet as possible about it …’1 With careful media management, some papers reported public sympathy for the family’s plight, but the republican press ‘argued that the disaffected working classes resented the hyperbole connecting the health of royal individuals with the political future of the entire nation.’1 The ensuing political manoeuvring meant that the public acquired an unusually deep understanding of ‘the royal disease’ ‒but arguably one that, by virtue of turning haemophilia into a political football, may have contributed to the stigmatisation that people experience to this day.
It is believed that haemophilia among British royalty emerged with Victoria’s father, Prince Edward, via a random mutation. In 2009, gene sequencing of bone samples of the Russian royal family identified the IVS3-3A>G mutation in the factor IX gene; this results in a causal substitution in the splice acceptor site of exon 4 ‒something that would cause severe haemophilia B.2,3
References
- Rushton AR. Leopold: the “bleeder prince” and public knowledge about hemophilia in Victorian Britain. J Hist Med Allied Sci 2012; 67: 457‒90. doi: 10.1093/jhmas/jrr029.
- Rogaev EI, Grigorenko AP, Faskhutdinova G, et al. Genotype analysis identifies the cause of the “royal disease”. Science 2009; 326: 817. doi: 10.1126/science.1180660.
- Green P. The ‘Royal disease’. J Thromb Haemost 2010; 8: 2214–5. doi: 10.1111/j.1538-7836.2010.03999.x.
1854
Florence Nightingale pioneers infection control read more
Florence Nightingale (1820–1910) – known as the Lady of the Lamp, a British icon, feminist, pioneer and founder of modern nursing ‒is widely regarded as a compassionate and brave hero in a time of exceptional brutality and suffering, and as a woman who achieved change in a world dominated by men. She abandoned a relatively privileged life against her family’s wishes to pursue a career as a nurse, obtaining the post of Superintendent at the Establishment for Gentlewomen During Illness in London’s Harley Street. In 1854, in response to public pressure, the Minister for War appointed her to lead a team of volunteer nurses to improve the dreadful state of military hospitals in the Crimea. Working at the barracks hospital in Scutari, Turkey, her reputation as a champion of the wounded and dying soldiers was forged.
But Florence Nightingale’s achievements in other fields should first be acknowledged. On returning to Britain, she successfully lobbied the Queen to set up a royal commission into the insanitary conditions suffered by the British Army. She was an eminent statistician and one of the first to present dry facts as accessible diagrams easily understood by the general public. With charitable funds raised in her name, she supported training for nurses at several London hospitals and was instrumental in transforming the profession’s mission and public standing. Her achievements were recognised with several public honours, and in 1907 she became the first women to be awarded the Order of Merit.
In 1858, Nightingale submitted to the Secretary of State for War her Notes on Matters Affecting the Health, Efficiency, and Hospital Administration of the British Army, Founded Chiefly on Experience of the Late War’.1With the benefit of hindsight, she summarises her evidence to the royal commission on the appalling sanitary conditions the soldiers endured during the Peninsular (1808–1814) and Crimean Wars (1853‒1856) and in army hospitals in Britain. It was never published but she circulated it privately. Her analysis showed that, on average, 39% of soldiers were sick at any one time. At one phase of the Crimean War, mortality due to disease was 73%. She wrote:
‘But if we go through the whole Crimean force, Regiment by Regiment, and mark what the Admissions into Scutari Hospitals were, and what the Deaths there during those fatal seven months, we shall see how much of the mortality was due in the Crimean case also to the frightful state of the General Hospitals at Scutari; how much it depended upon the number which each Regiment was unfortunately enabled to send to these pest-houses.’
Nightingale’s role at Scutari was primarily administrative, which probably explains her attention to effective provisioning and supply. She recognised that the sanitary conditions at the hospital were a contributory factor to the death rate, stating ‘want of ventilation, want of drainage, bad water, and foul air… nobody seems to have thought of trying the effect of pure air’. Despite Nightingale’s best efforts, mortality remained stubbornly high and it was not until a Government Sanitary Commission was sent to inspect the hospital that it was found to be built on a sewer. Nightingale had already noted how liquid faeces from patients with dysentery ran over the floors. When the sewer was flushed out and ventilation improved, the death toll began to fall.
Florence Nightingale is therefore widely seen as the pioneer who recognised that infection was a major cause of death during hospital care ‒but some say that her account of the importance of hygiene and sanitation was a late conversion. At Scutari, she had been more concerned with nutrition and the poor general health of soldiers, so that ‘She had helped them to die in cleaner surroundings and greater comfort, but she had not saved their lives.’2
During an inspection tour of hospitals in the Crimea, she became severely ill with brucellosis but insisted on continuing her task. The Queen was kept informed and there were prayers for her recovery. The Times christened her ‘The Lady of the Lamp’ and a ‘ministering angel’, capturing the sentiment of the day and defining her legacy. How Florence Nightingale came to be a powerful advocate of good hygiene and sanitation is perhaps beside the point. Her impact at the time was immense and her influence on the nursing profession is apparent over 150 years later.
Reference
- Nightingale F. Notes on Matters Affecting the Health, Efficiency, and Hospital Administration of the British Army, Founded Chiefly on Experience of the Late War. London: Harrison & Sons, 1858 (accessed March 2019).
- Bostridge M. Florence Nightingale: the Lady with the Lamp. Last updated February 2011. BBC History (accessed March 2019).
Further reading
McDonald L. Florence Nightingale, Nursing, and Health Care Today. New York: Springer Publishing,
2017. ISBN: 0826155588.
Baly ME, Matthew HCG. Florence Nightingale. Oxford Dictionary of National Biography. Version 06
January 2011. doi: 10.1093/ref:odnb/35241 (accessed March 2019).
1864
Louis Pasteur develops pasteurisation read
more
In the mid-19th century, the prevailing Western scientific opinion was that life was created spontaneously: put some nutrient broth in an open glass flask and, within a week or so, the mixture becomes cloudy with microbial growth that has generated spontaneously.
French chemist Louis Pasteur (1822–1895) did not accept this theory. In 1859, in the face of scepticism from the medical establishment, he ingeniously demonstrated that the source of microbes was contamination from the local environment.
This discovery was put to a practical test in 1863 when Napoleon III tasked Pasteur with solving a problem that was costing the French wine industry dearly: why did wine go off after being bottled? The principle of preserving food by boiling had been known for centuries, but this was not feasible for wine because it destroyed the flavour and evaporated the alcohol. Pasteur was able to show that it was necessary only to heat the wine to 50–60 degrees Celsius for a brief period to kill the causative organisms without significantly affecting the flavour. He had similar success with beer, and a grateful nation christened the process of heat treatment Pasteurisation in his honour.
Between 1865 and 1870, Pasteur’s research demonstrated that a blight threatening the French silkworm industry was due to an infection transmitted by parasites. On his advice, the infected worms were destroyed and another industry was saved. His subsequent work built on Jenner’s discovery of the principle of immunisation, developing attenuated vaccines against anthrax and rabies.
Pasteur carried out his work in the face of considerable personal tragedy: in 1868, a stroke left him partially paralysed and, between 1859 and 1866, he lost two daughters to typhoid and a third to cancer. The public health benefits of his treatment process began to be recognised by the early 1900s, but in the UK many people still contracted serious milk-borne infections. As late as 1943, it was known that 5%‒10% of the milk supply was contaminated by tuberculosis bacilli and 20%‒40% contained Brucella abortus. Many vociferously opposed milk pasteurisation because they thought its flavour and nutritional quality suffered. However, the gradual introduction of voluntary standards resulted in most of the milk supplied (to London, at least) being pasteurised by 1950.2
The UK still consumes more fresh milk than many other European countries, where heat-treated long-life milk is more acceptable. It is still legal to sell raw milk, provided it comes from herds that are registered free of tuberculosis and brucellosis and are regularly monitored, and only through designated outlets or direct online sales.3
References
- Wilson GS. The pasteurization of milk. Br Med J 1943; 1: 261‒2.
- Atkins PJ. The long genealogy of quality in the British drinking-milk sector. Historia Agraria 2017; 73: 35‒58. doi: 10.26882/HistAgrar.073E01a (accessed March 2019)
- Food Standards Agency. Raw drinking milk: advice on drinking raw milk and the safety regulations in place concerning its sale. Last updated September 2018 (accessed March 2019).
1865
George Mendel presents his work on genetic inheritance read more
In 1865, an Austrian monk presented the findings of his eight-year research project to the Society for the Study of the Natural Sciences in Brünn (Brno). In Experiments in Plant Hybridization, he described how he had grown approximately 10,000 pea plants and, from his observations, deduced how their characteristics passed between generations.
Gregor Mendel (1822‒1884) is now considered to be the father of modern genetics, but his research challenged scientific orthodoxy, provoked controversy and brought accusations of fraud. He did not live to see his work recognised by the scientific community.
Mendel was born in Moravia, then part of Austria and now in the Czech Republic. He was academically bright but his health was poor. After obtaining a degree in philosophy, in 1843 he joined a monastery, where he was able to continue his education. He studied agriculture and viniculture, and was ordained four years later. However, Mendel was unsuited to pastoral duties and failed to qualify as a teacher, so became a part-time assistant teacher. This allowed him the time and resources to pursue his scientific work and, between 1857 and 1864, he completed his study of hybridisation.
Accepted wisdom at the time was that heredity was the result of a blending process that diluted parental traits. Mendel observed that seven characteristics of peas showed no signs of dilution at all:
‘The angular wrinkled form of the seed, the green colour of the albumen, the white colour of the seed-coats and the flowers, the constrictions of the pods, the yellow colour of the unripe pod, of the stalk, of the calyx, and of the leaf venation, the umbel-like form of the inflorescent, and the dwarfed stem, all reappear in the numerical proportion given, without any essential alteration. Transitional forms were not observed in any experiment…’
Instead, he noticed that, in the first generation of plants, these characteristics appeared in the ratio three to one, labelling the prevailing characteristics ‘dominant’ and the latent ones ‘recessive’. Mendel had shown that inheritance depended on the combination of two unequally expressed genes, rather than a blend, thereby establishing two principles of heredity: the law of segregation (each gamete carries only one allele for each gene) and the law of independent assortment (genes segregate independently).
Mendel’s paper was largely ignored. Darwin’s On the Origin of Species by Means of Natural Selection, or the Preservation of Favoured Races in the Struggle for Life had been published in 1859, but it seems the potential link between the two theories was not made. European botanists confirmed Mendel’s work in 1900, but it was several more decades before it was accepted.
After being elected to succeed the abbot at the monastery in 1868, Mendel spent most of his time on administration, in particular opposing a new tax. He published only one more scientific paper, this time on hawkweed. However, it did not confirm his original hypothesis (because this plant reproduces asexually, generating clones) and was completely ignored. Mendel died, in considerable discomfort, of Bright’s disease (chronic nephritis), aged 62.
Further reading
Dunn PM. Gregor Mendel, OSA (1822–1884), founder of scientific genetics. Arch Dis Child Fetal Neonatal Ed 2003; 88: F537–9.
Bicknell R, Catanach A, Hand M, Koltunow A. Seeds of doubt: Mendel’s choice of Hieracium to study inheritance, a case of right plant, wrong trait. Theor Appl Genet 2016; 129: 2253‒66.
Gayon J. From Mendel to epigenetics: History of genetics. C R Biol 2016; 339: 225‒30. doi: 10.1016/j.crvi.2016.05.009.
1876
Alexander Graham Bell patents the telephone read more
It is rare that innovators develop their radical ideas in isolation. One example would be Gregor Mendel, who first described the pattern of heredity ‒and, for his trouble, was ignored by the scientific community until they caught up 35 years later. The development of the telephone is a case of the opposite: but for a phrase or two omitted from a patent application, the inventor uppermost in the public consciousness would be Antonio Meucci rather than Alexander Graham Bell. Both were working on electronic voice transmission; Bell got the patent.
Born in Scotland in 1847, Bell migrated to Canada in 1870 and moved to Boston in the United States the following year. He came from a family of elocution experts with a background in teaching deaf people a system of ‘visible speech’, and had been working on developing mechanical speech devices. He realised that sound waves could be converted into a fluctuating electric current which, if transmitted to a receiver, could be turned back into sound. By 1875 he had developed a basic device that could convert electricity into sound.
Bell had a powerful incentive to succeed. His patron was the lawyer and politician Gardiner Hubbard, whose daughter Mabel was deaf. Bell taught her to lip read and speak, then fell in love with her. Hubbard refused his consent to marriage until Bell had perfected his electrical speaking device. The prospective father-in-law was also an astute businessman and in 1876 filed a patent to protect the work. Crucially, this covered the electromagnetic transmission of vocal sound.
On 10 March 1876, Bell summoned his assistant from an adjacent room, shouting the words “Mr Watson, come here, I want to see you“, through his new telephone. The following year, Bell, Hubbard, Watson and others founded the Bell Telephone Company ‒Bell and Mabel were married two days later.
Coincidentally, an electrical engineer named Elisha Gray had also filed a patent for a telephone device on the same day, a matter of hours after that filed by Hubbard and at the same patent office. This prompted speculation that Bell’s application was granted only after some fancy footwork and resulted in the matter coming before the courts. The case was settled in Bell’s favour.
More seriously, an Italian American named Antonio Meucci had filed a temporary patent application in 1871 for his ‘Sound Telegraph’, a device he had been working on since 1849. He could not afford the fee for a full application and made no mention of electromagnetic voice transmission. The application expired in 1874 and he did not have the money to renew it. Meucci had shared a workshop with Bell in the 1870s. He sued Bell but died penniless in 1889 before the action was concluded. He was formally recognised by the US Congress as the true inventor of the telephone in 2002.
Bell fought several more court battles to defend his patent, eventually joining one litigant, Thomas Edison, to form the United Telephone Company. He continued his research in communication and techniques for teaching speech to the deaf, working with Helen Keller at one stage. He was one of the founding members of the National Geographic Society in 1888, and served as its second president from 1898 to 1903. He died on 2 August 1922.
Further reading
Alexander Graham Bell’s box telephone. National Museums Scotland. Available from https://www.nms.ac.uk/explore-our-collections/stories/science-and-technology/alexander-graham-bell (accessed March 2019).
Smith C. The history of the telephone: Six pioneers who transformed the world of communications. British Telecom. Updated 10 March 2018 (accessed March 2019).
Antonio Meucci. Famous Scientists: The Art of Genius (accessed March 2019).
1879
Thomas Edison invents the electric lightbulb read more
If you ever need a quote, Thomas Edison (1847‒1931) is your man. Google his name and up pop some very wise words, including:
I have not failed. I’ve just found 10,000 ways that won’t work
and:
Genius is one percent inspiration and ninety-nine percent perspiration
Those words certainly embody his life. Edison is described as America’s greatest and most brilliant inventor. He was certainly among the most productive, holding more than one thousand US patents. He is credited with inventing the phonograph (1877), the carbon telephone transmitter (used in telephones until the 1980s), a system for distributing electricity (though he was misguided in his criticism of alternating current), fluoroscopy (late 1890s), and was instrumental in developing the kinetoscope (1891) and an early battery (1910). But he is best known for developing the first commercially feasible incandescent electric light bulb in 1879. He achieved all this despite being profoundly deaf – something he said allowed him to concentrate.
Edison wasn’t the only inventor trying to develop a light bulb. However, he was the first to create a filament that could, when a voltage was applied to it, produce sufficient light without quickly burning up. His researchers tested thousands of carbonised plant materials and finally came upon cotton thread. In the high-quality vacuum he was able to create in bulbs drawn in his own glass blowing shed, the thread glowed for 15 hours before burning up. He refined the design until the bulb had a serviceable life, and then patented it in the United States.
As is the way with competitive inventors, the patent was disputed by a rival firm and it took six years for Edison to establish its validity. In turn, Edison sued the English inventor Joseph Swan, claiming infringement, but the two agreed to avoid a court battle by uniting their companies to form the Edison & Swan United Electric Light Company in Britain.
Edison didn’t invent the light bulb, but he created the first design that did the job. He then improved it again by using a carbonised bamboo filament that last 1,200 hours. The familiar modern version, with a spiral tungsten filament in a bulb filled with an inert gas, was introduced in 1913.
Further reading
Edison’s lightbulb. The Franklin Institute (accessed March 2019).
Lighting a revolution: U.S. patent 223,898, Thomas Edison’s incandescent lamp. Smithsonian Institution (accessed March 2019).
Thomas Edison. Wikipedia. Updated 11 March 2019 (accessed March 2019).
1887
Queen Victoria celebrates her Golden Jubilee read more
Royal jubilees are not entirely rare but certainly not common. There have been 66 monarchs of England or Great Britain since Athelstan in the tenth century, of whom six reigned for at least 50 years: Henry III, Edward II, James VI and I, George III, Victoria and Elizabeth II. Jubilees are known to have been celebrated in a significant way only since George III, who reigned from 1760 to 1820.
Victoria’s golden jubilee was celebrated on 20 and 21 June 1887. The official website of the royal family records that her day began with a quiet breakfast under the trees at Frogmore, the royal burial ground in Berkshire where her consort Prince Albert was buried. That evening, a royal banquet was attended by 50 foreign kings and princes, and the governing heads of Britain’s overseas colonies and dominions. The next day, blessed by unusually good weather, Victoria travelled to Westminster Abbey in an open carriage, greeted by cheering crowds all the way. She wrote in her journal:
‘No one ever, I believe, has met with such an ovation as was given to me, passing through those 6 miles of streets … The cheering was quite deafening & every face seemed to be filled with real joy. I was much moved and gratified.’1
The next day’s celebrations included meeting 10,000 schoolchildren in the rain on London’s Constitution Hill, followed by a civic reception in Slough.
The jubilee was marked by the kind of short-lived philanthropy that has been a feature of public celebrations over the years, the expense divided between the royal household and the state. Street feasts were provided for 400,000 of London’s poorest and another 100,000 in Manchester. There were firework displays and St Paul’s cathedral was illuminated by a son et lumière(sound and light) show; Nottingham, Bradford and Hull were awarded city status. Free ale and pipe tobacco were provided by the tea magnate Sir Thomas Lipton and the pubs stayed open until 2.30 am.
The golden jubilee marked the height of British global power and, as ever, no opportunity to promote the national interest could be ignored. Victoria had been crowned Empress of India in 1876 and the celebrations emphasised Britain’s interests in that country, with many Indian princes in attendance and the queen’s carriage led by an escort of Indian cavalry. But, although Victoria was a strong supporter of the empire and jingoism was common in political and social life, historians argue that Britain as a whole was not in the grip of imperialist fervour.2
The event also marked Victoria’s return to public life. She had rarely been seen in public since Albert’s death in 1861, after which wore only black (even at the jubilee), and a significant republican sentiment had emerged. However, Britain’s growing economic and imperial success bolstered her popularity in the 1870s and support for the queen was genuine among many people on the day. She celebrated her diamond jubilee with similar pomp and circumstance in 1897.
Victoria reigned until her death in 1901, having been head of state for 64 years during a remarkable century characterised by exceptional change.
References
- A history of jubilees. The Royal Household (accessed March 2019).
- Tombs R. Englishness in the English century. In: The English and their History. London, Penguin, 2014.
Further reading
Victoria (r. 1837-1901). The Royal Household (accessed March 2019).
Sully A. Queen Victoria and Britain’s first diamond jubilee. BBC News. 22 May 2012 (accessed March
2019).
Queen Victoria’s golden jubilee. Making Britain. Discover how South Asians shaped the nation,
1870-1950. The Open University (accessed March 2019).
1895
Wilhelm Röntgen produced and detected x-rays read more
The German physicist Wilhelm Conrad Röntgen was awarded the 1901 Nobel prize in physics for his discovery of x-rays. His breakthrough arose during research into the properties of the recently discovered cathode rays ‒the technology that would eventually give the world television.
Röntgen was born in the Lower Rhine Province of Germany in 1845; his family moved to the Netherlands when he was three. Following a largely unremarkable school career – marked only by an aptitude for making mechanical devices ‒he studied physics at university, where his academic star began to shine. He was strongly influenced by the work of August Klundt and Rudolf Clausius on thermodynamics and light; in 1869, after graduating with a PhD from the University of Zurich, Röntgen worked with Klundt for several years.
Several professorships in European cities followed: Röntgen was, it seems, head-hunted by the top universities of the day until, in 1900, he accepted the Chair of Physics at the University of Munich, where he remained for the rest of his career.
During the late 19th century, several scientists had been investigating the rays emitted by the cathode when an electrical current passed through a gas held at low pressure in a glass discharge tube. It was known that the rays could be deflected by electrical and magnetic fields, and they were subsequently found to have the properties of subatomic particles later known as electrons. The cathode ray tube, which utilises a vacuum rather than a low pressure gas, provided the image in the first television in 1926 and was used in sets until the advent of plasma, LCD and OLED screens in the early 21stcentury.
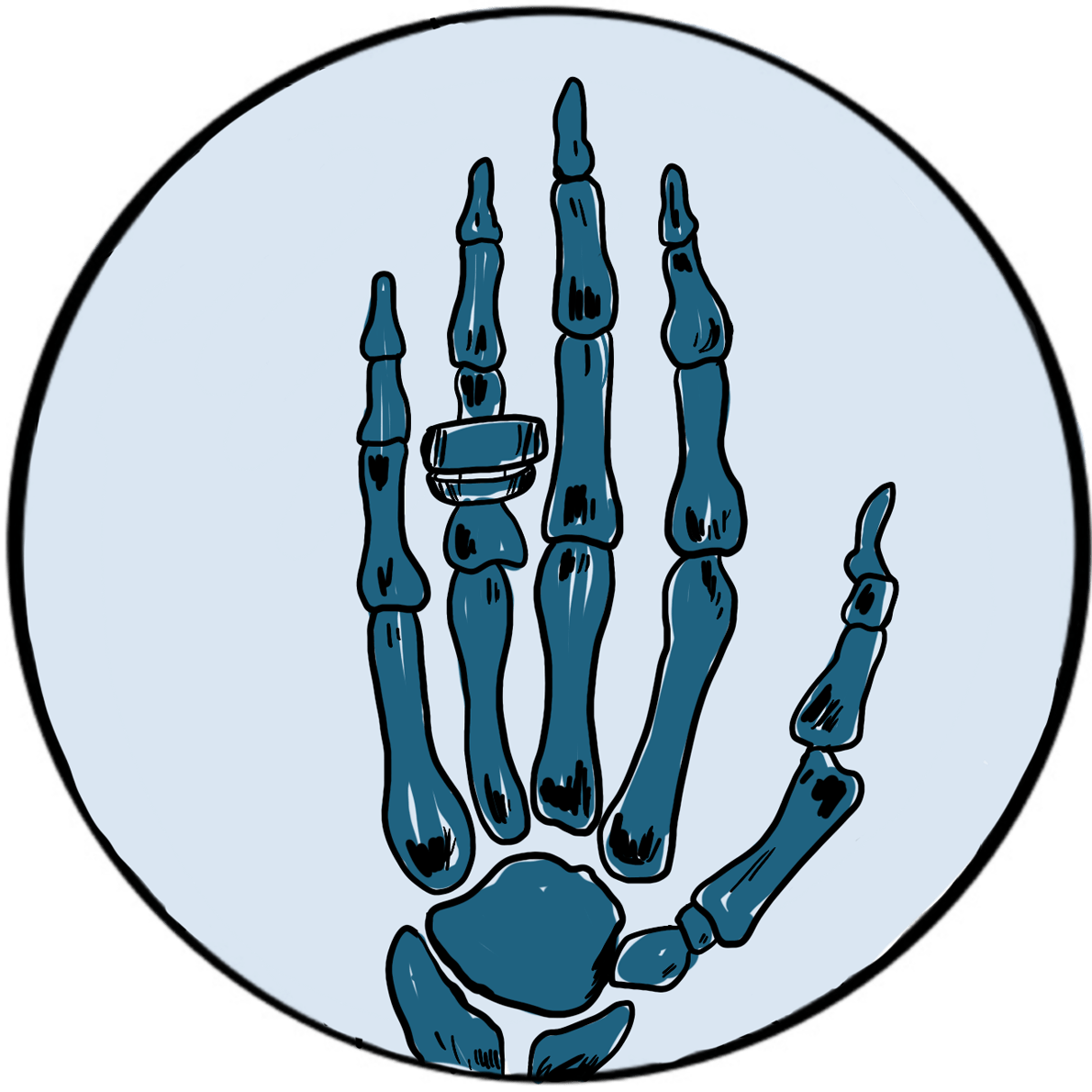
Röntgenobserved that if the discharge tube was sealed to exclude light, a plate coated with barium platinocyanide (a compound that emits light when exposed to radiation) fluoresced when placed in the path of the rays in a darkened room. He subsequently showed that putting various objects in the path of the rays attenuated the fluorescence to varying degrees but did not prevent it. He then placed his wife’s hand between the discharge tube and a photographic plate; when this was developed, he had an image of the hand, with dark shadows cast by the bones, faint shadows from the flesh and the clear outline of a ring. This was the first Röntgenogram. When his wife saw the image, she is said to have proclaimed “I have seen my own death!”.
Although he realised that the rays producing these images were not the same as the cathode rays in the discharge tube, Röntgen was uncertain of their nature and christened them x-rays. They were later shown by Max von Laue to be electromagnetic waves with properties similar to light ‒ a discovery for which he was awarded the Nobel Prize for Physics in 1914.
X-rays were used as a diagnostic tool at the turn of the century, though machines were cumbersome and unreliable. In 1911, US President Theodore Roosevelt was shot in an unsuccessful assassination attempt; x-rays were used to locate the bullet, showing that the risk of moving it was greater than leaving it be.
Although he was showered with honours for his research, Röntgen is said to have been a modest and reticent person; he was colour blind and nearly blind in one eye. Preferring to work alone, he had built much of his apparatus himself. He gave the money from his Nobel prize to his university. He died in 1923 from bowel cancer.
Further reading
Wilhelm Conrad Röntgen: biographical. The Nobel Prize (accessed March 2019).
Gunderman RB. X-Ray Vision: The Evolution of Medical Imaging and Its Human Significance. Oxford University Press, 2013. ISBN-13: 978-0199976232.
Howell JD. Early clinical use of the x-ray. Trans Am Clin Climatol Assoc 2016; 127: 341‒9.
1898
Radium is discovered by Pierre and Marie Curie read more
In 1896, the French physicist Henri Becquerel was one of many scientists working on Röntgen’s newly discovered x-rays. Having inherited some uranium salts from his father, he was trying to determine whether there was a link between the naturally phosphorescent substance and x-rays. He noted that uranium was capable of fogging a photographic plate when placed nearby, suggesting that the salts emitted rays, and went on to show that (unlike x-rays) these rays could be deflected by an electromagnetic field and could ionise gas. Becquerel had discovered spontaneous radioactivity, for which he was awarded the 1903 Nobel prize in physics. The cause of his death in 1908 is unknown, although he is believed to have experienced serious burns from handling uranium.
Becquerel shared his Nobel prize with Marie and Pierre Curie for their work on spontaneous radiation. Pierre was born in Paris in 1859. After enrolling at the Sorbonne, he rose through the ranks of academia to become Professor of General Physics in the Faculty of Sciences. Marie was born in Poland in 1867 and had a distinguished academic career, joining the Sorbonne in 1891. They were married in 1895.
The Curie’s research on radioactive substances proved to be ground-breaking, but was carried out under poor conditions and supported by a heavy teaching commitment. They were the first to use the term ‘radioactivity’ to describe this newly discovered property of emitting rays. They found that pitchblende (now called uraninite) was more radioactive than uranium and must therefore contain one or more other radioactive substances. In 1898, their development of techniques to refine pitchblende led to the discovery of two rare elements: radium and polonium. Pierre and Marie were both enthusiastic advocates of therapeutic applications of radioactivity; Radium Institutes were later established in Paris, London and Warsaw to develop medical uses.
The Nobel prize was originally awarded only to Pierre, but he insisted his wife also be cited in recognition of her work. In 1906, he fell under a horse carriage and was killed. Marie succeeded him as Professor of General Physics at the Sorbonne, the first woman to hold the position. In 1911, she was awarded the Nobel prize in chemistry for the discovery of radium and polonium, and for research into the properties of radium.
Both Pierre and Marie experienced radiation poisoning. Marie died of aplastic anaemia in 1934. Their laboratory books are still too radioactive to handle without protection. Their daughter Irene, also a physicist, and her husband Frédéric Joliot-Curie were also both eminent scientists and were awarded the 1935 Nobel prize in chemistry for the discovery of artificial radioactivity; both probably died of radiation poisoning.
Units of radioactivity were named after Becquerel and the Curies in their honour. A becquerel is defined as one disintegration per second; a curie (now discontinued) is 3.7 x 1010 becquerels.
Further reading
Marie Curie: biographical. The Nobel Prize (accessed March 2019).
Pierre Curie: biographical. The Nobel Prize (accessed March 2019).
Marie and Pierre Curie: a marriage of true minds. Marie Curie Blog. 10 February 2015 (accessed March 2019).
1900-1949









1901
US Surgeon General Catalogue lists treatments for haemophilia read more
The dawn of the 20thcentury shed little light on how best to treat haemophilia. Medical and scientific journals had published several accounts of generations of families affected by haemophilia and, when treatment was mentioned, most had reported great difficulty – or failure – in controlling bleeding.
In 1803, for example, the Philadelphia physician John Otto listed bark, astringents (topically and internally), strong styptics, opiates and oral sulphate of soda (sodium sulphate) as the measures employed to stem bleeding.1 Only sodium sulphate proved effective – a finding that others confirmed.
In 1840, the London physician Samuel Lane recorded how he had tried to promote the formation of a clot by applying ‘gum tragacanth and beaver gathered from a hat’.2 Success was temporary: the clot was soft and broke away with renewed bleeding.
It is thanks to the efficiency of the army and its enthusiasm for record-keeping – or that of its librarians, at least ‒ that we have a picture of the remedies in vogue at the turn of the century. The 1901 index-catalogue of the library of the Surgeon-General’s Office, United States Army, lists the publications about haemophilia held by the library.3 The titles reveal the variety of interventions that doctors considered worth reporting. They include lime (calcium oxide and/or hydroxide), oxygen, thyroid gland or extract, injection of serum, hydrogen peroxide, gelatin (including by injection), suturing, compression, calcium chloride and bone marrow.
The London medical historian G.I.C. Ingram observed that more treatments were listed in the following years, some topical to promote clot formation, others systemic.4 They include injection of sodium citrate, calcium lactate or of Witte’s peptone (a protein hydrolysate), anaphylaxis, splenectomy, the ‘galvanic needle’ (passage of electric current via a wire), animal and human serum, adrenaline, the ‘muscle jostles’ of a bird, x-rays, transfusion of the patient’s own blood, oral tissue fibrinogen, female hormones, oxalic acid, a ‘bromide extract of egg white’ and the venom of Russell’s viper (which promotes thrombosis by activating factor X). Ingram notes that viper venom was the only treatment to undergo systematic evaluation. Robert MacFarlane, a junior demonstrator of pathology at London’s St Bartholomew’s Hospital (who would go on to describe the enzyme cascade underpinning coagulation and to identify haemophilia B), and Burgess Barnett, curator of reptiles at the Zoological Society of London, showed that a very dilute solution of the venom stopped bleeding immediately when applied topically.5
Although the first successful use of blood transfusion to treat bleeding due to haemophilia was published in 1840,2 it was not until 1926 that the Surgeon General’s catalogue included publications documenting its use.
References
- Otto JC. An account of an hemorrhagic disposition existing in certain families. Reprinted from The Medical Repository and Review of American Publications on Medicine, Surgery and the Auxiliary Branches of Science 1803, vol. 6, 1‒4. Clin Orthop Relat Res 1996; 4‒6 (accessed 26 February 2019).
- Lane S. Haemorrhagic diathesis. Successful transfusion of blood. Lancet 1840; 35: 185‒8. doi: 10.1016/S0140-6736(00)40031-0.
- Library of the Surgeon-General’s Office (US). Index-catalogue of the Library of the Surgeon-General’s Office, United States Army. Authors and subjects. Series 2, vol. 6. Washington: GPO, 1880‒1932 (accessed March 2019)
- Ingram GIC. The history of haemophilia. J Clin Path 1976; 29: 469‒9 (accessed March 2019).
- Macfarlane RG, Barnett B. The haemostatic possibilities of snake venom. Lancet 1934; 2: 985‒7. doi: 10.1016/S0140-6736(00)43846-8.
1914 - 18
World War I read more
In 1914, there existed 194 treaties providing arbitration for international disputes and 400 peace organisations worldwide ‒ 46 in Great Britain and 72 in Germany. In 1913, the Dutch had opened a Palace of Peace to host treaty conferences. Peace was in abundance, but no one was surprised when the British Empire declared war on Germany at 11 pm on 4 August.
Europe festered with rivalry and bitterness. Since 1900, Germany had invested heavily in its navy to match British sea power, but was running out of money. France, Russia and Britain signed alliances despite overt rivalry in the expansion of their colonies. As early as 1908, a German military, beyond government control, had developed the Schlieffen Plan for a rapid invasion of Belgium, then defeating France before turning on Russia. In 1912‒13, smaller countries aligned to one side or the other ‒ Serbia, Bulgaria, Romania, Greece, Montenegro and Turkey ‒ were at war. The Balkans were an open sore between Austria-Hungary and its ally Germany, and Russia, supported by France.
On 28 June, Archduke Franz Ferdinand, heir to the thrones of Austria and Hungary, and his wife Sophie, were murdered by a Serb-trained assassin, lighting the fuse to what should have been, but never was, the ‘war to end all wars’. Both sides believed they could win, though Germany feared its military power would be outstripped by France unless it acted. It implemented the Schlieffen Plan, overrunning Belgium and declaring war on France on 3 August. Britain saw no choice but to side with France.
We look back in horror at what followed, but at the time the war was seen – in Britain at least – as a fight for freedom and a better world. Even so, there was little by way of gung-ho enthusiasm at the prospect of war. Despite the ‘Your Country Needs You’ propaganda, those who could remember the events of the American Civil War of 1861– 865 and the Boer War of 1899–1902 understood that the traditional tactics of trench warfare and cavalry charges were suicidal in the new era of mechanical weaponry.
By a quirk of the Gregorian calendar, 1916 provided an extra day for futile slaughter: 29 February was the ninth day of the Battle of Verdun, a ten-month campaign by the German military specifically to tie down and destroy the French army. Verdun, a town near the border with Belgium and Germany, was strongly fortified and came to represent French national pride; there would be no surrender. In June, the Russians launched an offensive that would divert German military resources, and further north, in July, the British and French began the Battle of the Somme. The German army was eventually forced to withdraw from Verdun to meet these challenges.
The casualty rate at Verdun is not certain, but is estimated to have totalled between 650,000 and 750,000, of whom perhaps 300,000 men were killed or missing. No ground was won or lost, though the battle was probably decisive in shoring up French resolve.
In the 1,560 days of the war, nine million soldiers were killed in Europe alone, including one third of all British men who were aged 19–22 when the conflict started. Douaumont, one of the defensive forts at Verdun, is now a memorial and the site of an ossuary (www.verdun-douaumont.com) housing the remains of 130,000 soldiers from the battlefield.
1918
Tsarevich Alexis dies read more
Often described as the most famous person with haemophilia, Alexei Nikolaevich, the Tsarevich of the Russian empire at the beginning of the 20thcentury, had the misfortune to be born into the royal Romanov family at a time when inherited wealth and privilege became distinctly unpopular.1
Alexei was born in 1904, the son of Alexandra Feodorovna, a German aristocrat and the granddaughter of Queen Victoria, and Tsar Nicholas II. He was their fifth child and first son ‒ and, as late-arriving heir to the crown, bore the considerable weight of the empire’s hope and expectation on his shoulders. Alexandra, as a result of her descent from the British royal family, was a carrier of haemophilia. She had not been a popular choice for Nicholas and, with the risk of haemophilia well known at the time, the marriage was opposed by others in the royal family.
It was apparent shortly after birth that Alexei had haemophilia. This would have been perceived by the population as a judgement from God for some failing of the ruling class, and every attempt was made to keep the fact secret. He grew up cosseted and protected, but many accidents and bleeding episodes are documented, at least one of which resulted in Alexei receiving the last rites. Partly to cope with this, Alexandra removed herself from public life, increasing her unpopularity.
Conventional medicine had little to offer, and it is believed this was one factor behind the family’s attachment to Grigori Rasputin. The peasant healer is reputed to have achieved several miraculous cures when Alexei was ill, including one via a telegram sent from Siberia. Rasputin’s closeness to the family and his growing political influence caused resentment among the aristocracy, resulting in his murder in 1916.
In 1917, as Russia suffered during the First World War, public unrest stoked by food rationing culminated in the February Revolution. The army sided with the Bolsheviks and Nicholas was forced to abdicate. The new government wanted him to hand power to Alexei, who they believed would be easily controlled. However, Nicholas knew the boy was not strong enough and abdicated in favour of his less pliable brother, Michael. For his part, Michael knew he could not keep power and soon abdicated himself.
The family were imprisoned in Yekaterinburg, a city about 1,600 miles from St Petersburg, until April 1918, when victory by the anti-Bolshevik White Army threatened. Alarmed by the possibility that the royals might be reinstated, the government ordered their execution. It was a brutal and bloody affair: the family had sewn jewels into their clothing for safe-keeping, which prolonged the carnage by deflecting some of the bullets and bayonets. Even the 13-year-old boy with haemophilia wouldn’t die until he was shot in the head.
The bodies were buried before the White Army took the city eight days later. The remains were discovered in 2007 and identified by DNA testing a year later. It was then confirmed that Alexei had haemophilia B, just like the other affected royals.2
It is arguable that haemophilia was instrumental in the demise of the Russian royal family.1Alexei’s haemophilia contributed to the Romanovs’ close relationship with Rasputin and to Alexandra’s withdrawal from public life; and it influenced Nicholas’s decision not to hand power to his son.
References
- Brown A. The Royal Disease and The Royal Collapse: Political Effects of Hemophilia in the Royal Houses of Europe. 2017. Student research. 63. DePauw University (accessed February 2019).
- Green P. The ‘royal disease’. J Thromb Haemost 2010; 8: 2214–5. doi: 10.1111/j.1538-7836.2010.03999.x.
1926
The first description of Von Willebrand disease is published read more
In April 1924, a five-year-old girl called Hjördis attended a clinic at the Deaconess Hospital in Helsinki, Finland. She came from the Åland Islands, situated about halfway between Helsinki and Stockholm, Sweden. She had a history of severe bleeding from the nose, after tooth extraction and from the ankle, and had been hospitalised for ten weeks at age three due to bleeding from a deep wound in her lip.
Hjördiswas one of 11 children, of whom seven experienced excessive bleeding and three had died. The physician she had seen at the clinic was the hospital’s physician-in-chief, Dr Erik Adolf von Willebrand, a well-regarded doctor with a long interest in haematology and in blood clotting in particular. He found that Hjördis’s blood count was normal, as was her clotting time (an indicator of thrombin formation); she had slight anaemia and slight thrombocytopenia. But, unusually, her bleeding time (which depends on vasoconstriction and platelet function) was two hours – at least ten times longer than expected.Von Willebrand concluded that her disorder was the result of platelet dysfunction, combined with a defect of the vascular wall.
Von Willebrand obtained the co-operation of a local teacher to document the family history, and discovered that the bleeding disorder affected 23 of 66 family members. By contrast with haemophilia, the disorder affected both women and men. Bleeding did not affect muscles and joints, but mostly the mucosae, causing epistaxis, oral bleeding, bruising, heavy menstrual bleeding and excessive bleeding after childbirth. His findings, published in 1926, described a disorder he labelled pseudohaemophilia.Hjördis died from menstrual bleeding, aged 13.
Von Willebrand wasn’t the first to describe a bleeding disorder affecting both women and men – the earliest account seems to have been in 1852, though the cases were labelled haemophilia. However, he provided the best early scientific description, and went on to collaborate with other Scandinavian physicians and publish distinguished research. In recognition of this work, the disorder was named von Willebrand disease between the late 1930s and early 1940s, by which time more cases had been reported.
It was not until 1971 that the disorder was attributed to dysfunction of a large protein that acts as a carrier of factor VIII; the protein was christened von Willebrand factor (VWF). Further research in the 1970s and 1980s identified several subtypes of von Willebrand disease, characterised by different molecular defects. The VWF gene was cloned in 1985.
Further reading
Berntorp E. Erik von Willebrand. Thromb Res 2007; 120: S3–S4. doi: 10.1016/j.thromres.2007.03.010.
Federici AB, Berntorp E, Lee CS. The 80th anniversary of von Willebrand’s disease: history, management and research. Haemophilia 2006; 12: 563–72. doi: 10.1111/j.1365-2516.2006.01393.x.
1928
Penicillin discovered by Alexander Fleming read more
In 1929, Alexander Fleming, a 48-year-old microbiologist working at St Mary’s Hospital, London, published On the antibacterial action of cultures of a penicillium, with special reference to their use in the isolation of b. influenzae.1
Fleming’s paper described his observation a year earlier that bacterial growth in culture plates was inhibited by the presence of a contaminant mould, Penicillium. He is said to have been an untidy worker and had left the plates exposed to the air when he went on holiday.
Reasoning that the mould was secreting an antibacterial substance into its environment, Fleming tested other moulds for similar activity and found none. He prepared a filtrate from a broth culture of Penicillium and determined its solubility, stability to heat and its activity after dilution. He named the filtrate ‘penicillin’ and found it inactive against some bacteria, including the enterococci and Haemophilus influenzae, but highly active against Gram-positive bacteria including staphylococci and streptococci. He tested its toxicity in mice and the human eye and found it safe, concluding: “It is suggested that it may be an efficient antiseptic for application to, or injection into, areas infected with penicillin-sensitive microbes.”
Fleming was not the first to describe the antibacterial activity of a mould.2It had long been known that some moulds had antiseptic properties. In the second half of the 19thcentury, several accounts from Europe and South America reported that Penicilliuminhibited bacterial growth. In 1897, French PhD student Ernest Duchesne presented a thesis in which he showed that injecting a Penicillium broth into guinea pigs protected them against infection; his work was ignored. Fleming himself acknowledged the work of the Belgian microbiologist AndréGratia, who had published his research on the antibacterial activity of Penicilliumin 1924 but had been unable to pursue his research due to ill health.
Even after Fleming’s paper, development of the therapeutic applications of penicillin was slow: it was difficult to produce in quantity and Fleming was more interested in exploring its physicochemical properties. Australian-born Howard Florey, chair of pathology at Oxford University, and the German-born biochemist Ernst Chain repeated Duchesne’s work in 1938 and began to produce small quantities of penicillin.3,4 They successfully treated the first patients in 1941.5
It was not possible to scale up production of penicillin in wartime Britain, so Florey and Chain travelled to the United States where, with the support of John Fulton, a professor of physiology at Yale University, the large scale process was developed. Florey declined to patent the production process.
Fleming, Florey and Chain shared the 1945 Nobel prize in physiology or medicine “for the discovery of penicillin and its curative effect in various infectious diseases.”
References
- Fleming A. On the antibacterial action of cultures of a penicillium, with special reference to their use in the isolation of b. influenzae. Br J Exp Pathol 1929; 10: 226–36 (accessed March 2019).
- Arseculeratne SN, Arseculeratne G. A re-appraisal of the conventional history of antibiosis and penicillin. Mycoses 2017;60: 343–7. doi: 10.1111/myc.12599.
- Kyle RA, Steensma DP, Shampo MA. Howard Walter Florey ‒ production of penicillin. Mayo Clin Proc 2015; 90: e63-e64. doi: 10.1016/j.mayocp.2014.12.028.
- Chain E, Florey HW, Gardner AD, et al. Penicillin as a chemotherapeutic agent. Lancet 1940; 236: 226‒8. doi: 10.5555/uri:pii:S0140673601087281.
- Abraham EP, Chain E, Fletcher CM, et al. Further observations on penicillin. Lancet 1941; 238: 177‒89. doi: 10.5555/uri:pii:S0140673601087281.
1939 - 45
World War II read more
The major economies of the world enjoyed a short period of prosperity and sociopolitical enlightenment in the years immediately following the end of hostilities in 1918‒but it was a future built on shaky foundations. The Allies had imposed the Treaty of Versailles on an unwilling Germany, many of whose people were not reconciled to their defeat in the war of 1914–1918. Britain and France were struggling to meet the costs of their victory, but the reparations provided for in Versailles were not adequate and, worse, fuelled further resentment in Germany. The United States had bankrolled Europe’s war effort and its defensive trade policy made it difficult for the old order to repay its debts.
American avarice and regulatory disinterest brought down the global economy with the Wall Street Crash in 1929. European countries lacked the resilience to weather the storm, none more vulnerable than Germany, which relied on the US loans now being recalled. The result was bank failures, unemployment and mass hunger. Over the next four years, successive German administrations tried and failed to restore the economy, ultimately resorting to the introduction of government by decree. Faith in democracy waned and the electorate turned to extremist parties – communist and fascist – for solutions. The fascists prevailed.
Active since the early 1920s, the Nazi Party had established support among the wealthy, the middle class and the rural population. Its leader, Adolf Hitler, portrayed as the country’s saviour, found scapegoats everywhere, but particularly targeted the Jewish population and other minorities as a threat to Aryan purity and economic prosperity. Hitler became chancellor in 1933, purging his party of dissenters and banning political opposition. He abandoned the Treaty of Versailles and reintroduced conscription. When President Hindenberg died in 1934, Hitler declared himself president, chancellor and head of the army – with the title of Führer – and the army and judiciary swore an oath of allegiance to him, not the state.
Europe looked on, impotent. Enlightened politicians hoped Hitler’s ambitions could be contained by diplomacy and appeasement. Fear of war was uppermost, and there were Nazi sympathisers in government and among the aristocracy. The pope struck a deal with the Nazis so that they and the Catholic church would not interfere with each other’s affairs. Lloyd George, Britain’s former leader, called Hitler ‘the greatest German of the age’.
In 1936, German troops illegally entered the Rhineland, then occupied by France. Britain was less than sympathetic to its ally and the intrusion went unchallenged. Hitler exploited the Spanish Civil War of 1936–1939 to test his army and air force., and strengthened links with the Italian fascist leader Mussolini. In 1938, Germany annexed Austria and was allowed to annex Sudetenland in Czechoslovakia in return for signing the Munich Agreement, which aimed to guarantee peace. In November of that year, the Nazis destroyed Jewish shops and synagogues throughout Germany, an event to become infamous as Kristallnacht.
Germany continued to expand into neighbouring countries during 1939, breaching the Munich Agreement. On 31 March, Britain and France pledged to guarantee the independence of Poland. Germany agreed a non-aggression pact with the Soviet Union to divide Eastern Europe. On 1 September, Hitler invaded Poland (as did the Soviet Union) and precipitated war in Europe. In June 1941, Germany opened a second front against the Soviet Union, beginning a four-year conflict that cost the lives of millions.
Failing to contain Nazi aggression, the Allies turned to the United States for help. Though the American administration had provided non-combative support, the nation was split on whether to get involved, with some maintaining that the war was a European affair. Everything changed on 7 December 1941, when the Japanese air force bombed the unsuspecting American fleet in Pearl Harbour, its base in Hawaii. On 8 December 8, the United States and Great Britain declared war on Japan; on 11 December, Germany and Italy declared war on the United States. The second great war of the twentieth century was now truly global, bringing to the world the Holocaust, chemical and nuclear weapons, and the deliberate mass killing of civilians by bombing from the air.
War in Europe ended on 8 May 1945 and in the Pacific on 15 August, after the United States dropped two atomic bombs on Japan. It is estimated that 20 million military personnel and 40 million civilians died in the war. The world is struggling to cope with its economic and political legacy to this day.
1947
Haemophilia A and haemophilia B are shown to be different read more
In 1947, Alfredo Pavlovsky (1907–1984), a haematologist from Buenos Aires, Argentina, reported that a mixture of blood from two people with haemophilia had a shorter coagulation time than unadulterated blood from either individual.1
For some time, there had been a suspicion that haemophilia was not a single entity, but Pavlovsky is credited with the first recorded observation leading to the recognition of two types of haemophilia: one due to a deficiency of factor VIII, later labelled haemophilia A; the other, less common, caused by a deficiency of factor IX, and first known as Christmas disease before being relabelled haemophilia B.
When Pavlovsky combined blood from a person with haemophilia A and from another with haemophilia B, the mixture contained sufficient factor VIII and factor IX to clot normally. However, he did not interpret his findings in this way. Pavlovsky believed that haemophilia was likely to be caused by an excess of ‘anticoagulant substances’ rather than ‘a deficit in the globulin fraction’, though he did not exclude that possibility, or that the two might coexist.
Haemophilia B was formally identified as a distinct disorder from haemophilia A in 1952. It was initially labelled Christmas disease, after the first patient described.2
References
- Pavlovsky A. Contribution to the pathogenesis of haemophilia. Blood 1947; 2: 185‒91 (accessed March 2019).
- Biggs R, Douglas AS, MacFarlane RG, et al. Christmas disease: a condition previously mistaken for haemophilia. Br Med J 1952; 2: 1378‒82 (accessed March 2019).
1948
The NHS is founded on 5th July read
more
The 1946 National Health Service Act came into effect on 5thJuly 1948. Its purpose was to ‘provide for the establishment of a comprehensive health service for England and Wales, and for purposes connected therewith.’ Separate legislation covered Scotland and Northern Ireland.
The Act stated: ‘it shall be the duty of the Minister of Health to promote the establishment of a health service to secure improvement in the physical and mental health of the people and the prevention, diagnosis and treatment of illness.’
The 1942 Beveridge Report provided the foundation for what was to become the welfare state. It conceived a system of social insurance ‘from cradle to grave‘, funded by taxation. The unemployed, sick, retired and widowed would receive benefits, and there would be an acceptable minimum standard below which no one would be allowed to fall. The proposals proved popular and, following a landslide victory by the Labour party in the 1945 general election, the transformation of British society began. The first Minister of Health was Aneurin Bevan.
The NHS was born out of the sense of collective social responsibility that emerged during and after the Second World War. Previously, healthcare had been provided privately or in local authority voluntary hospitals and asylums. Since 1911, all male employees had received healthcare paid for by national insurance contributions, but this did not extend to their families. In some parts of the country, medical aid societies were established and funded by local workers. They proved very successful and were the inspiration for Bevan’s NHS.
Services previously provided by voluntary organisations and private practitioners were brought under one roof and provided free at the point of use. Local authorities were given responsibility for community services, including the national immunisation programme, maternity clinics and community nurses.
Not everyone was inspired by the prospect of utopia. Doctors didn’t want to become state employees and threatened to boycott the NHS unless Bevan recognised their independent status and allowed their private practice to continue.1 In 1948, the NHS budget was £437 million (equivalent to £15 billion today). Uptake of the new service exceeded expectations and the Labour government soon introduced charges. The legislation needed to allow prescription charges was passed in 1949; charging for prescriptions – originally one shilling per prescription, then one shilling per item ‒dental services and spectacles began in 1952.2 Successive governments have wrestled with the challenge of the cost of the welfare state ever since. The 2018/19 NHS budget was £115 billion, of which charges account for 1.2% (about £1.4 billion).3
As of 2019, the NHS is arguably the last element of the welfare state to have survived anything like intact. The Labour governments of the 1990s introduced private providers to widen choice for patients, increase capacity and promote competition. The role of the private sector is now seen as beneficial,4 and it is acknowledged that the welfare state model has been replaced by mixed provision by private and publicly funded services.5
The NHS currently employs 1.5 million people and provides some aspect of care for one million people every 36 hours. The OECD estimates that the UK spends about 9.2% of gross domestic product on health, ranking it ninth and exactly average in Europe.6 It still delivers comprehensive care, largely free, but in recent years the UK has not compared well with other large economies in key health markers such as cancer outcomes, infant mortality and avoidable mortality.7
References
1. British Medical Association. People’s History of the NHS (accessed March 2019).
2. House of Commons Health Committee. NHS Charges. Third Report of Session 2005–06. Volume I.
Report, together with formal minutes. Ordered by The House of Commons to be printed 6 July 2006
(accessed March 2019).
3. The King’s Fund. How the NHS is funded. May 2017 (accessed March 2019).
4. The King’s Fund. Is the NHS being privatised? October 2018 (accessed March 2019).
5. The National Archives. Citizenship. Brave new world: The welfare state (accessed March 2019).
6. OECD/EU. Health at a Glance: Europe 2018. State Of Health In The Eu Cycle. Paris: OECD
Publishing, 2018. Revised version, February 2019. doi: 10.1787/health_glance_eur-2018-en (accessed March 2019).
7. Robertson R. How does the NHS compare internationally? The King’s Fund. June 2017 (accessed March 2019)
Further reading
National Health Service Act, 1946. An act to provide for the establishment of a comprehensive
health service for England and Wales, and for purposes connected therewith. Chapter 81. 6th
November 1946 (accessed March 2019).
NHS. About the NHS. April 2016 (accessed March 2019).
1950
The Haemophilia Society is established as a charity read more
The Haemophilia Society is the UK’s largest charity representing everyone affected by a bleeding disorder ‒ not only haemophilia, and not only people with the bleeding disorder, but their families and carers too. Its aim is: ‘to provide easy access to information and opportunities, influence national policy and practice to make the care and treatment of bleeding disorders consistent, effective and accessible to all and enable the voices of all people with bleeding disorders to be heard.’
Haematologist GIC Ingram has described how the Society came into being thanks to the sociability of people with haemophilia. He records how, during the 1940s, Frank Smith and Bob White started chatting about haemophilia while waiting for their clinic appointments at a London hospital. Together with four others who attended the same clinic, they began to meet informally at the Francis Galton Laboratory for National Eugenics. In 1942, together with others with an interest in haemophilia, they formed the International Haemophilia Society (IHS) and began to reach out to more people with haemophilia. The new society remained informal until the post-war years, when a channel of communication was opened up with Sir Weldon Dalrymple-Champneys, deputy chief medical officer at the Ministry of Health. With his support, the IHS registered as a charity with the London County Council in 1950.
The IHS was soon holding its committee meetings at the Hospital for Sick Children in Great Ormond Street, London, beginning a close relationship that survives to this day. Its position was consolidated over the next few years and, in 1954, the IHS worked with the Ministry of Health on establishing haemophilia centres (beginning with the Oxford centre), and addressing the challenges faced by people with haemophilia in education and employment, and with transport.
The IHS was reconstituted and renamed the Haemophilia Society in 1954, and local groups were formed throughout the country. Close contact with the Ministry of Health was maintained and Sir Weldon became the Society’s president in 1957.
Part of the Society’s purpose was to raise awareness about haemophilia – however, it faced a ‘chicken and egg’ problem when it came to fundraising as few people knew about bleeding disorders. Its first big boost came from the actor Richard Burton who, after visiting the Oxford haemophilia centre, pledged the proceeds from the royal premiere of his 1969 film Where Eagles Dare to the appeal that carried his name, raising a much-needed £4,000 (equivalent to well over £60,000 in 2019).
The Society played a significant part in establishing the World Federation of Hemophilia in the 1960s, and has been instrumental in the campaign to persuade the UK government to pay compensation to people with haemophilia who were infected with HIV by contaminated blood. Today, the Haemophilia Society is still run largely by volunteers and continues to vigorously promote awareness and support research. It campaigns on behalf of people with bleeding disorders and has a prominent role in the current public inquiry into the infected blood scandal. Membership is free and open to everyone.
Further reading
The Haemophilia Society. Available at https://haemophilia.org.uk (accessed March 2019).
Ingram GI. The history of haemophilia. J Clin Path 1976; 29: 469‒79.
1950-2000








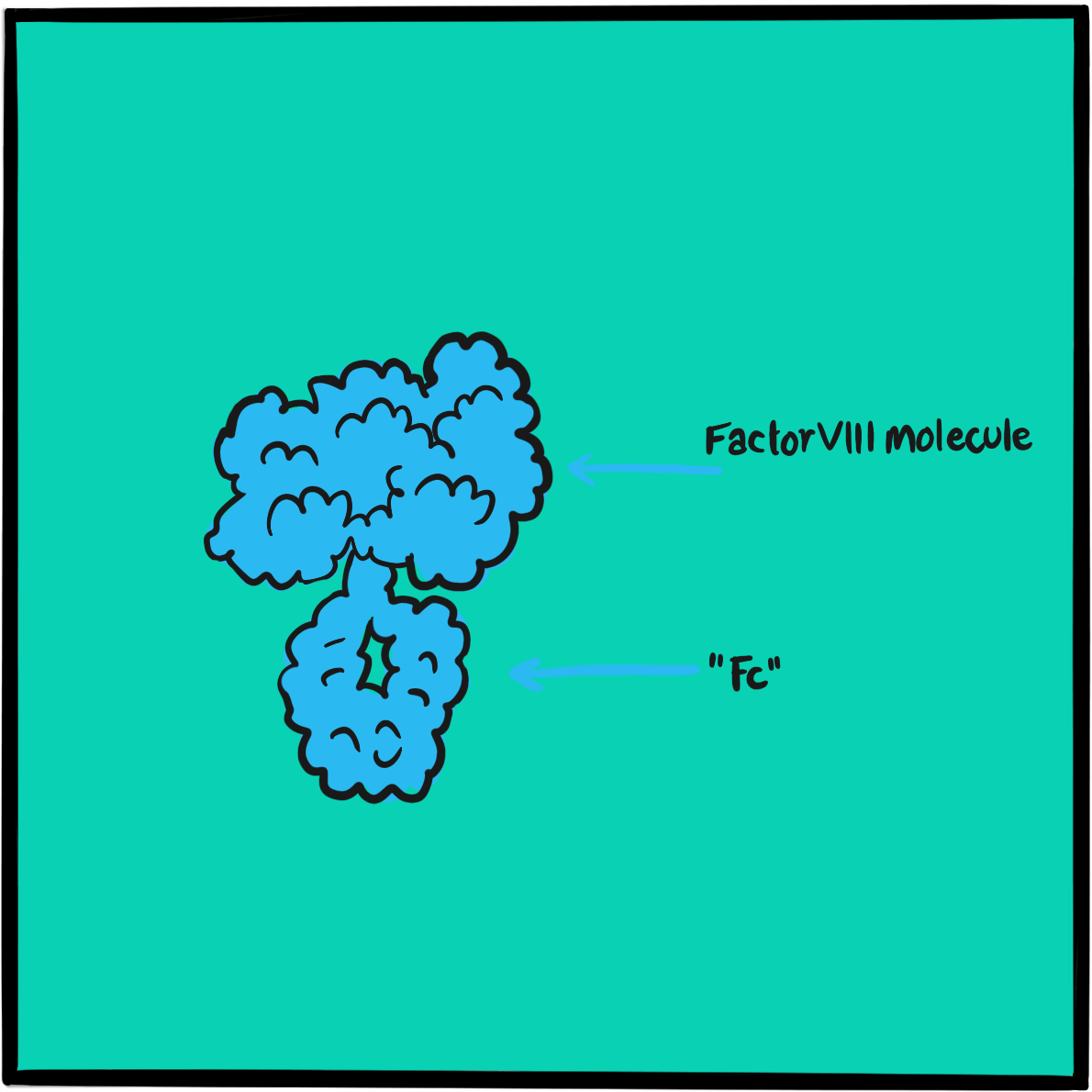





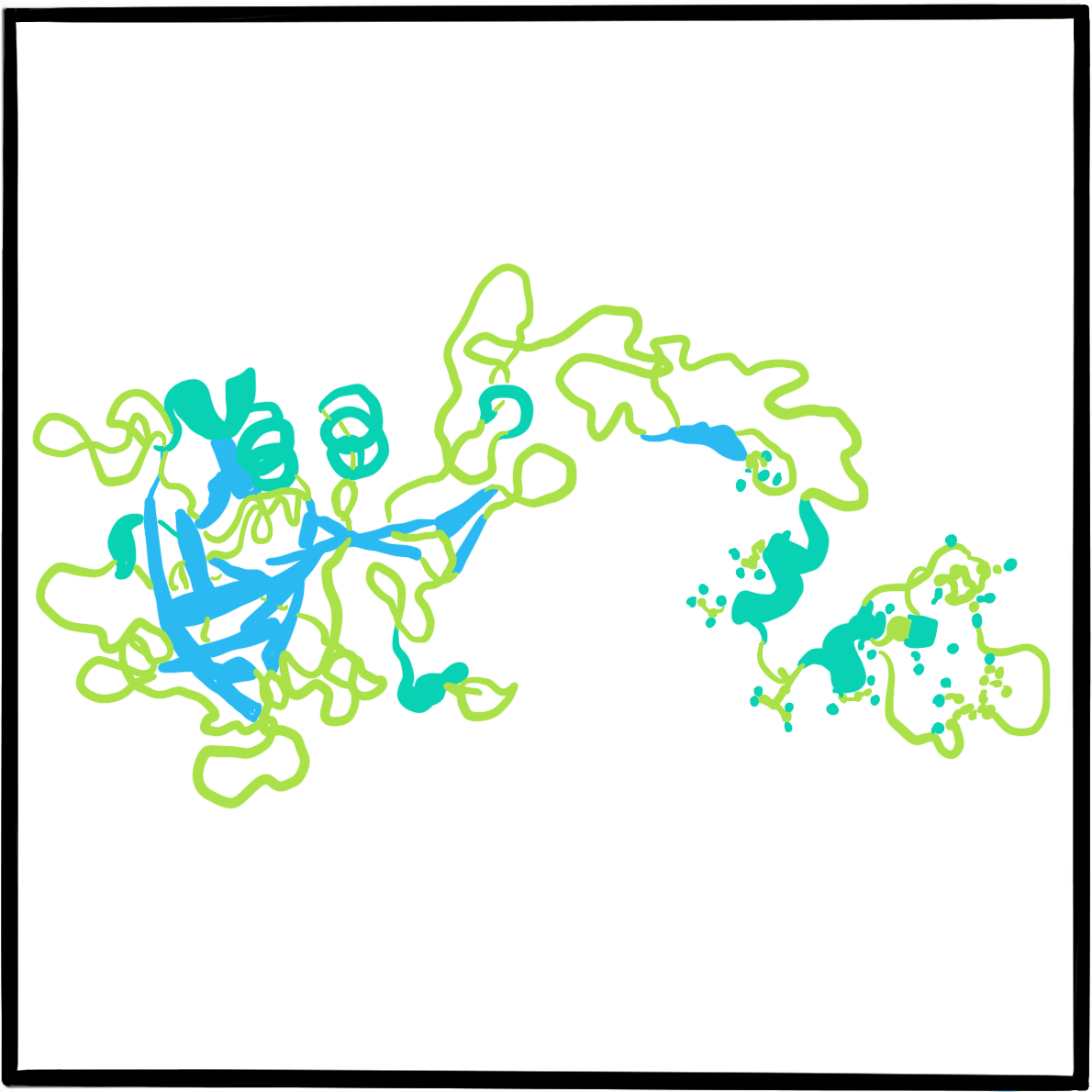





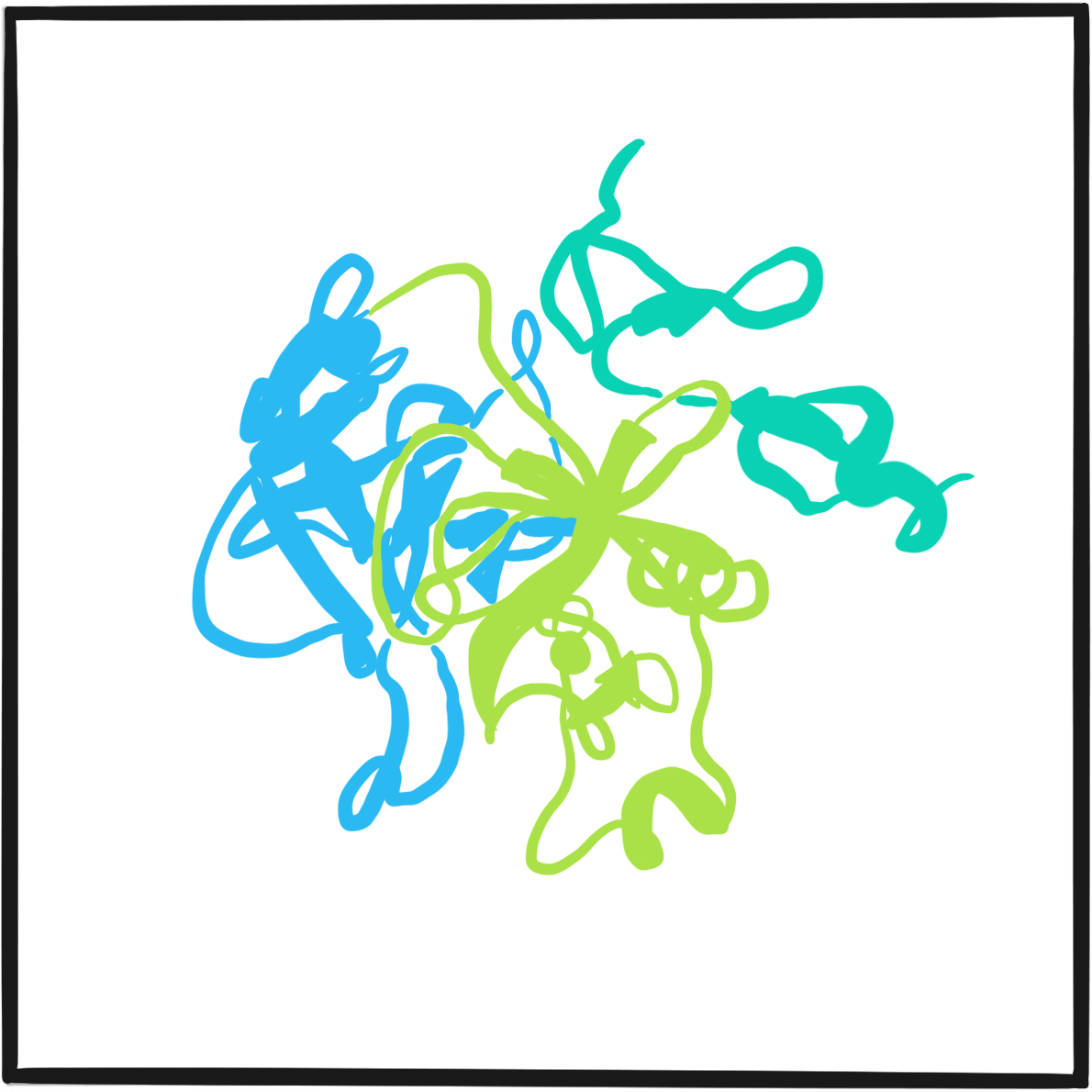
1952
Identification of Christmas Disease (Haemophilia B) read more
The name ‘Christmas disease’ was coined in 1952 by a team of haematologists led by Rosemary Biggs from the Radcliffe Infirmary, Oxford.1In a paper published in the British Medical Journal, they described seven people who appeared to have haemophilia but whose blood clotted more quickly when mixed with that from other people with haemophilia. They mentioned five earlier publications documenting the same phenomenon, including that of Argentinian haematologist Alfredo Pavlovsky, who first reported evidence of more than one type of bleeding disorder in people diagnosed with haemophilia.2
The first person Biggs and her colleagues described was Stephen Christmas, a five-year-old boy from London. At that time, haemophilia was believed to be caused by a failure or delay in blood thromboplastin formation caused by absence or deficiency in the blood of ‘antihaemophilic globulin’ (now known as factor VIII). If that was true, then Christmas disease must be a different disorder. Biggs’s team correctly identified a strong functional association between the ‘Christmas factor’ they suspected was deficient in their newly identified disorder and factor VII. Their work explained why, by contrast with most people with haemophilia, bleeding in a person with Christmas disease could not be treated by infusion of antihaemophilic globulin. It was surely no coincidence that their paper was published in the Christmas edition of the British Medical Journal.
Stephen Christmas contracted HIV from transfused blood in Canada and died from the complications of AIDS in 1993. Rosemary Biggs went on to have an illustrious career in haematology under the mentorship of Robert MacFarlane, who first described the coagulation cascade. In 1955, they co-authored the first British guideline on the treatment of haemophilia.3
References
- Biggs R, Douglas AS, MacFarlane RG, et al. Christmas disease: a condition previously mistaken for haemophilia. Br Med J 1952; 2: 1378‒82 (accessed March 2019).
- Pavlovsky A. Contribution to the pathogenesis of haemophilia. Blood 1947; 2: 185‒91 (accessed March 2019).
- MacFarlane RG, Biggs R. The diagnosis and treatment of haemophilia and its related conditions. Memo Med Res Counc 1955; 32: 1‒23.
1953
Crick, Watson, Wilkins and Franklin work out the structure of DNA read more
The 1962 Nobel prize in physiology or medicine 1962 was awarded to Francis Crick, James Watson and Maurice Wilkins ‘for their discoveries concerning the molecular structure of nucleic acids and its significance for information transfer in living material’.
Reprehensibly, the prize failed to acknowledge the contribution of the x-ray crystallographer Rosalind Franklin. Popular understanding of how the structure of DNA was discovered has been coloured by Watson’s singular and chauvinistic memoir, The Double Helix (1968), in which his description of Franklin was ‘exceedingly distasteful: cruel, misogynist, and flippant’. Watson later admitted his mistake.1
DNA was first described in 1871 by the Swiss chemist, Friedrich Miescher. He had been trying to identify the proteins in the nucleus of white blood cells, but realised the new substance was fundamentally different; he called it ‘nuclein’. His discovery was largely ignored.
In 1919, the Russian biochemist Phoebus Levene proposed that nucleic acids (as they were now known) were made up of nucleotides. However, he could not work out how they were arranged to form the molecule. Canadian-American physician Oswald Avery showed in 1944 that DNA was the source of genetic code. In 1950, Austrian biochemist Erwin Chargaff then demonstrated that bases in DNA occur in pairs and that the base composition of DNA varies between species.
The search for the structure of DNA turned into a race. In Britain, Cambridge researchers Watson and Crick were working with x-ray crystallographers Franklin and Wilkins from London, spurred on by competition from American scientist Linus Pauling (though Watson was also American). Franklin worked in Wilkins’ department, but they had a sour relationship that seems to have interfered with them fully understanding the potential of their work.
It was Franklin’s measurements of the molecule that enabled Watson and Crick to generate what proved to be a remarkably accurate model of DNA. They published their work in April 1953, accompanied by Franklin’s structural description.2,3 By this time, Franklin had left Wilkins’ department to work in another laboratory. She died of ovarian cancer four years later.
References
- McCrum R. The 100 best nonfiction books: No 15 – The Double Helix by James D Watson (1968). The Guardian. 9 May 2016 (accessed March 2019).
- Watson JD, Crick FH. Molecular structure of nucleic acids: a structure for deoxyribose nucleic acid. Nature 1953; 171: 737‒8 (accessed March 2019).
- Franklin RE, Gosling RG. Molecular configuration in sodium thymonucleate. Nature 1953; 171: 740–1 (accessed March 2019).
Further reading
Cobb M. Life’s Greatest Secret: The Story of the Race to Crack the Genetic Code. London: Profile Books, 2015.
Pray LA. Discovery of DNA structure and function: Watson and Crick. Nature Education1(1): 100 (accessed March 2019).
1958
Inga Marie Nilsson develops the concept of prophylaxis read more
By the late 1950s, the effectiveness of treating bleeds in people with haemophilia A with antihaemophilic fraction (AHF) was well established. AHF, which contained factor VIII, was a stable preparation (albeit crude by modern standards) prepared by sequentially precipitating the proteins in human plasma. One dose required up to 1.6 L of blood and contained 3 g of a mixture of proteins that included clotting factors. AHF was given on demand to treat bleeding episodes and to prevent bleeding during surgery.
In 1958, the Swedish haematologist Inga Marie Nilsson and her colleagues Margarita Blömback and Olof Ramgren described three boys with severe haemophilia A who were the first to be treated using a new approach: prophylaxis.1 They showed that regular doses of AHF provided sufficient clotting factor to lower the risk of bleeding from severe to moderate, and this reduced the number of days spent being treated in hospital; in one boy it also prevented overt joint damage. The boys received half a dose of AHF once a month for 3.5 years, which maintained their own ‘AHF’ level at 1%‒3% almost until the next dose. The authors concluded: ‘Prophylactic treatment of haemophilic patients may be of value in decreasing the frequency of severe bleeding episodes.’
Nilsson reported further experience of prophylaxis after up to 13 years and up to 25 years.2,3 By this time, modern factor VIII preparations were being given in a three times weekly regimen and the approach was used for people with severe haemophilia A or B. Nilsson’s thorough long-term follow-up studies provided the strong evidence on which current management strategies are based. She showed that initiating prophylaxis at an early age (1–2 years) to maintain factor VIII activity above 1% meant ‘it is almost possible to prevent bleedings and haemophilic arthropathy’ compared with a control group treated on demand. In 1992, following Nilsson’s account of 25 years’ experience, a commentator expressed concerns about the risk of poor adherence and the high cost of prophylaxis, but conceded ‘we must seriously entertain the possibility of a change in our treatment philosophy.’4
There was a downside. Although prophylaxis was remarkably well tolerated, five of the 66 people in Nilsson’s study developed inhibitors, 50 were infected with hepatitis C, 11 were infected with HIV and one died of the complications of AIDS.3
Inga Marie Nilsson had an illustrious career: she developed a treatment for inhibitors, contributed to the identification of von Willebrand disease (VWD) as a disorder distinct from haemophilia, and described genetic variants of VWD, as well as conducting research in other aspects of coagulation and thrombosis. She died in 1999 after a short illness, aged 66.
Reference
- Nilsson IM, Blömback M, Ramgren O. Haemophilia in Sweden. VI. Treatment of haemophilia A with the human antihaemophilic factor preparation (fraction I–0). Acta Med Scand Suppl 1962; 379: 61‒110. doi: 10.1111/j.0954-6820.1962.tb18848.x.
- Nilsson IM, Hedner U, Ahlberg A. Haemophilia prophylaxis in Sweden. Acta Paediatr Scand 1976; 65: 129‒35. doi: 10.1111/j.1651-2227.1976.tb16525.x.
- Nilsson IM, Berntorp E, Löfqvist T, Pettersson H. Twenty-five years’ experience of prophylactic treatment in severe haemophilia A and B. J Intern Med 1992; 232: 25‒32. doi: 10.1111/j.1365-2796.1992.tb00546.x.
- Aledort LM. Prophylaxis: the next haemophilia treatment. J Intern Med 1992; 232: 1‒2. doi: 10.1111/j.1365-2796.1992.tb00543.x.
1960
FXIII identified read more
In 1960, Swiss haematologist François-Henri Duckert and his colleagues described a family affected by ‘a congenital haemorrhagic diathesis with slow and poor wound healing’.1 Those affected rarely bled spontaneously but did so after trauma; bleeding onset was delayed by 24–36 hours but then persisted for several weeks. Clots formed but were ‘remarkably loose’ and healing was slow. The disorder affected both sexes and was probably inherited in an autosomal recessive pattern.
Duckert suggested the cause was probably a deficiency of fibrin stabilising factor, a component of the coagulation system proposed in 1944, confirmed in 1948 by Hungarian biochemists Kalman Laki and Laszlo Lóránd, and subsequently known as factor XIII (FXIII).2,3
It is now known that FXIII is activated by thrombin and calcium ions to form cross-links between fibrin strands, strengthening a clot and stabilising it by slowing the degradation of fibrin by the anticoagulation system. It is a complex entity consisting of two pairs of proteins, A2and B2. It has many physiological functions in addition to its role in the coagulation system, including the formation of new blood vessels, endothelial cell proliferation and barrier function, modulation of the activity of cells involved in inflammation and promotion of atherosclerosis, and the development of bone.4
Laszlo Lóránd recounts his involvement, as a research student, in the discovery of FXIII deficiency with an enthusiasm only a scientist could muster:
‘Few biological phenomena evoke a more dramatic effect in an audience than seeing a large volume of Ca2+‐chelated plasma turn into a clot by adding a drop of thrombin. As if by magic, the liquid is suddenly converted into a gel that can no longer be poured out of a flask, and it is immediately recognized that clotting would be life‐saving at wound sites, but could be deadly within the circulation.’5
Over 20 years later, Lóránd and his colleagues developed a quantitative test for FXIII that enabled geneticists to confirm Duckert’s view of the pattern of inheritance. FXIII is very rare, with a prevalence of 1–3 per million population (though regional differences are marked). To date, more than 100 gene mutations causing FXIII deficiency have been identified.6
References
- Duckert F, Jung E, Shmerling DH. A hitherto undescribed congenital haemorrhagic diathesis probably due to fibrin stabilizing factor deficiency. Thromb Diath Haemorrh 1960; 5: 179‒86.
- Robbins KC. A study of the conversion of fibrinogen to fibrin. Am J Physiol 1944; 142:581‒8. doi: 10.1152/ajplegacy.1944.42.4.581.
- Laki K, LórándL. On the solubility of fibrin clots. Science 1948; 108: 280. doi: 10.1126/science.108.2802.280.
- Schroeder V, Kohler HP. New developments in the area of factor XIII. J Thromb Haemost 2013; 11: 234‒44. doi: 10.1111/jth.12074.
- Lóránd L. Factor XIII and the clotting of fibrinogen: from basic research to medicine. J Thromb Haemost 2005; 3: 1337‒48.
- de Moerloose P, Schved JF, Nugent D. Rare coagulation disorders: fibrinogen, factor VII and factor XIII. Haemophilia 2016; 22 Suppl 5: 61‒5. doi: 10.1111/hae.12965.
1963
The World Federation of Haemophilia is established read more
The World Federation of Hemophilia (WFH) was established in 1963 by Frank Schnabel, a Canadian businessman with severe haemophilia A. He was a founding member of the Canadian Hemophilia Society in 1953 and had seen it grow from a small support group in Montreal to a national voluntary organisation. By the early 1960s, patient-led organisations had developed in many countries and, working with representatives from twelve national patient associations (Argentina, Australia, Belgium, Canada, Denmark, France, Germany, Japan, the Netherlands, Sweden, the UK and the USA), Schnabel convened the inaugural meeting of the WFH in Copenhagen, Denmark, on 25 June 1963. The Federation was formally constituted as a charity in 1964, with Schnabel as its first chair.
From the outset, Schnabel’s ambition was to create an organisation that could represent people with haemophilia the world over, and support collaboration between health professionals, researchers and people with haemophilia and their families. To promote an international network, raise awareness and share knowledge, the WFH initiated the bi-annual WFH congresses, which to this day are among the premier events in the scientific calendar for the haemophilia community. In 1969, the WFH was officially recognised by the World Health Organization.
It has long been clear to everyone in the haemophilia community that the most effective treatments are not equally accessible to everyone around the world. The WFH established the International Hemophilia Treatment Centre Program to help share knowledge between member societies in economically developed and developing countries through outreach training and fellowships.
Frank Schnabel died in 1987. Alan Tanner succeeded him as acting chair of the WFH, and Brian O’Mahoney was elected president at the WFH General Assembly in 1994. O’Mahoney renewed the Federation’s commitment to help redress the imbalance in access to modern treatments through initiatives including the a twinning programme between haemophilia centres in developed and economically developing countries. In Chile, the first ‘winning coalition’ of government support, pharmaceutical companies (which donated medicines) and the national patients’ organisation proved to be an effective model for capacity building and improving haemophilia care. This model has since been successfully implemented elsewhere.
Building on the success of the winning coalition concept, the WFH launched its Global Alliance for Progress (GAP) Program in 2003. This ten-year initiative put into effect all that the WFH had learned about building sustainable care in 20 economically developing countries, with the aim of tackling under-recognition of haemophilia by raising the number of people diagnosed to 50,000 ‒a figure more in line with the known prevalence of the disorder. GAP was followed by the Cornerstone Initiative, which is focusing resources on economically developing countries where standards of care are least advanced. Cornerstone currently serves eight countries in Africa, South and East Asia and Southeast Asia and the Western Pacific.
WFH now has 140 member organisations and is supported by all the major pharmaceutical companies involved in the development and provision of treatments for haemophilia. Although it is now an international organisation that commands respect on the global stage, it still relies on the efforts of volunteers to deliver change at a local level.
Further reading
World Federation of Hemophilia. About us. Updated June 2018 (accessed March 2019).
1963
The Beatles have their first UK no. 1 read
more
Things weren’t going very well for The Beatles at the end of 1962. Their first single, Love Me Do, was virtually ignored by their record label and by the BBC, which had an almost complete monopoly on music radio at the time. It was a repetitive three-chord ditty with fewer than 20 different words. The single wasn’t even played by The Beatles proper: producer George Martin had been dissatisfied first with drummer Pete Best and then his replacement Ringo Starr, to the extent that he replaced Ringo with a session drummer for the recording. Manager Brian Epstein had secretly bought 10,000 copies of the record because he’d been told that was the sales figure required for a top twenty hit. The song reached number one on Merseyside but crawled in at number 17 on the national chart.
During the sessions for Love Me Do, the soon-to-be moptops had recorded Please Please Me. The early version was slow and hadn’t inspired George Martin, but when he recorded a new up-tempo rendition he declared, ‘Gentleman, you have just made your first Number One.’
In 1963, Britain was enduring its worst winter weather for years. Please Please Me was released in January and broadcast on ITV’s Thank Your Lucky Stars to a snowbound population glued to Saturday night TV. Within three weeks, it was in the top twenty and, according to some music papers, it soon made number one. Although the all-important ‘official’ national chart only placed it at number two, it marked the beginning of the transformation of pop music.
The single’s reception meant that an LP was now required, and The Beatles recorded their first album, also titled Please Please Me, in only 16 hours. Released in March 1963, the LP reached number one in the album chart in May and stayed there for six months (until it was pushed off by The Beatles’ second LP, With The Beatles). To coincide with the LP, the group released a third single: From Me To You also reached number one in the official UK chart in May 1963. Beatlemania was now in full swing and the next single, She Loves You, topped the charts within days of its release in September. It was knocked off the number one spot only by I Want to Hold Your Hand. By the end of the year, it was estimated The Beatles had sold seven million records.
Further reading
Norman P. Shout! The True Story of The Beatles. London: Elm Tree Books, 1981.
Tobler J. The Beatles. London: Deans International Publishing, 1984.
The Beatles. The Beatles Anthology. London: Cassel & Co., 2000.
1964
Robert Macfarlane describes the coagulation cascade read more
“After years of confusion, it seems that a relatively simple pattern is emerging from present theories of blood coagulation.”
So wrote Robert Gwyn MacFarlane in 1964, in a letter to the scientific journal Nature titled An enzyme cascade in the blood clotting mechanism, and its function as a biochemical amplifier.1
It was indeed relatively simple but, much like a user’s manual for an electronic gadget, only when you understand it in the first place. Researchers had been struggling for years to work out how the various components of blood interacted to form a clot. MacFarlane described how a succession of studies stretching back to 1953 had revealed the various stages of coagulation. He summarised this in a simple diagram in which the proteins (i.e. clotting factors) were elegantly shown to be activated in a sequence of eight steps, each dependent on its predecessor.
MacFarlane described the cascade as a series of enzyme-proenzyme transformations, each regulated by positive and negative feedback mechanism. This explained how a single event could precipitate a rapid and marked increase in blood coagulability (the ‘amplifier’) and why coagulation did not continue to expand beyond the site of injury.
The cascade is very close to the current (slightly more complex) model of coagulation. The main differences are that MacFarlane included factors V and VIII as if they acted independently; they are now considered to be cofactors when in their activated forms (that is, they act in combination with another factor). Factor VIII, which is deficient in people with haemophilia A, is a cofactor for activated factor IX (which is deficient in haemophilia B); together they form a complex that contributes to the conversion of factor X to Xa.
MacFarlane died in 1987. He was a pathologist at Oxford’s John Radcliffe Hospital, and part of the team that first described haemophilia B in 1952, having carried out pioneering work on coagulation by studying snake venom. In 1988, the British government set up the Macfarlane Trust in his name to support people with haemophilia infected with HIV by contaminated NHS blood products. Its duties were taken over by the NHS in 2017 and its assets were transferred to the Terence Higgins Trust, which continues this work.
References
- MacFarlane R. An enzyme cascade in the blood clotting mechanism, and its function as a biochemical amplifier. Nature 1964; 202: 498‒9. doi: 10.1038/202498a0.
- Biggs R, Douglas AS, MacFarlane RG, et al. Christmas disease: a condition previously mistaken for haemophilia. Br Med J 1952; 2: 1378‒82 (accessed March 2019).
1964
Comprehensive care for bleeding disorders begins read more
The management of haemophilia requires more than treatment for acute bleeding. It involves the prevention of bleeding and joint damage; prompt management of bleeding; management of complications (including joint and muscle damage and other sequelae of bleeding, inhibitor development, viral infection transmitted through blood products); and attention to psychosocial health.1 All this is delivered by a package of services and resources known as comprehensive care.
The World Federation of Hemophilia defines comprehensive care as a process that ‘promotes physical and psychosocial health and quality of life while decreasing morbidity and mortality… to provide or coordinate inpatient (i.e. during hospital stays) and outpatient (clinic and other visits) care and services to patients and their family.’1It is provided by a multidisciplinary team, the core of which comprises a medical director, a nurse coordinator, a musculoskeletal expert (e.g. a physiotherapist), a laboratory specialist, and a psychosocial expert (such as a social worker or psychologist). This team, which should be experienced in the management of bleeding disorders, calls on a wide range of other specialties when needed.
Comprehensive care represents the best model of care that a well-resourced health system can offer to people with a bleeding disorder and their families. An approach that was developed during the 1960s, two of the most prominent clinicians involved were Netherlands paediatrician Simon van Creveld (1894–1971)2 and UK haematologist Katharine Dormandy (1926–1978).3
Van Creveld became director of the paediatric clinic at the University of Amsterdam in 1938. He retired from this post in 1964, having contributed significantly to research into clotting mechanisms at a haemophilia clinic he had founded near Amsterdam. In that year, the clinic was officially re-opened by Queen Juliana of the Netherlands as the first comprehensive treatment centre for people with haemophilia. Van Creveld said: ‘With the opening of the Hemophilia Clinic at Huizen, in 1964, the Netherlands became the first country in the world where hemophilic children could receive comprehensive treatment and be educated to take their place in the outside world.’
The centre subsequently focused on clinical work and, now affiliated with the University of Utrecht, it is known as the Van Creveld Kliniek. Van Creveld’s pioneering work influenced a generation of health professionals and established the primacy of the comprehensive care model in The Netherlands.
Katharine Dormandy was already an experienced researcher and an advocate of the emerging network of haemophilia centres in the UK when, in 1964, she joined the Royal Free Hospital in London. She established a haemophilia centre serving the North East Thames NHS region (in a caravan in the car park) where, for the first time in the UK, a full-time nurse provided on-demand factor infusions. Dormandy was a pioneer of holistic care for haemophilia, and helped to develop education resources for boys with haemophilia. In 1966, she wrote: ‘It is not enough for young haemophiliacs merely to survive, nor even for them to grow to manhood with good physique – they must also be able to take their place in society as well-integrated, independent individuals, able to earn a salary and support their families.’4
In 1970, Dormandy introduced a programme of home treatment with cryoprecipitate. By this time, with support from the UK Haemophilia Society, the centre had expanded and now delivered a comprehensive care programme in collaboration with other specialties at the hospital. In 1977, Katharine Dormandy was the first recipient of the RG Macfarlane Award, conferred by the Haemophilia Society for her outstanding contribution to the social and physical wellbeing of people with haemophilia and related disorders. The Royal Free centre was renamed the Katharine Dormandy Haemophilia and Thrombosis Centre in 1978.
References
- World Federation of Hemophilia. WFH Guidelines for the Management of Hemophilia. 2nd edition. London: Blackwell, 2012. Available from http://www1.wfh.org/publications/files/pdf-1472.pdf (accessed May 2019).
- Van Mourik JA. Van Creveld, pioneer of hemophilia care and coagulation research in the Netherlands: a personal account. J Thromb Haemost 2004; 2: 1029‒33.
- Hawgood BJ. Rosemary Biggs MD FRCP (1912-2001) and Katharine Dormandy MD FRCP (1926-78): from laboratory to treatment and care of people with haemophilia. J Med Biogr 2013; 21: 41‒8. doi: 10.1258/jmb.2011.011041.
- Britten MI, Spooner RJD, Dormandy KM, Biggs R. The haemophilic boy in school. Br Med J 1966; 2: 224‒8.
Further reading
Nathwani A. Celebrating 50 years of haemophilia care at the Royal Free Hospital. J Haem Pract 2015; 2: 1-2. doi: 10.17225/jhp00047.
1965
Professor Judith Graham Pool prepares cryoprecipitate from plasma read more
Physicians in the 19thcentury had shown that blood transfusion would correct the coagulation defect in people with haemophilia, a property believed to be due to the action of platelets. Transfusion offered an effective acute treatment for severe bleeding; however, due to the large volume of blood needed to provide sufficient clotting factor, it was not feasible for routine use.
It became clear during the 1930s that plasma was as effective as whole blood in stopping bleeding. The active component was one or more of the proteins that could be precipitated from plasma.1,2 Initially, the precipitate was obtained by acidification, but by the mid-1940s it had been shown that this could also be achieved by freezing and slow thawing ‒though this had to be done with care because its clotting activity was readily lost. The protein mixture prepared in this way was termed ‘cryoprecipitate’.
In 1955, Emile Rémigy, a PhD student at the University of Nancy in France, showed that a plasma preparation concentrated by freezing could correct the coagulation defect when infused in people with haemophilia A as well as a larger volume of normal human plasma.3 However, as his research was published in French it remained unknown to the English-speaking scientific community.
Work continued on cryoprecipitate, but research was hampered by the challenges of producing a sterile preparation in sufficient quantities. In 1961, a commercial preparation became available in the United States, marketed as a source of fibrinogen.
In 1959, Judith Graham Poole, a physiologist at California’s Stanford University, had developed an assay for factor VIII. When assaying frozen plasma fractions, she and co-worker Jean Robinson knew the importance of slow thawing; however, when they tested the resulting liquid they found little FVIII activity. On testing the sludge left at the bottom of the container, they discovered why: the precipitate contained FVIII in a concentration 50 times greater than in plasma. In 1964, Poole reported the first successful use of cryoprecipitate to treat four people with haemophilia, noting that an injection of no more than 50 ml could raise clotting factor activity to 40% of normal.4
Poole realised she and Robinson had found a method for extracting FVIII from plasma that could be used by local institutions such as blood banks, and went on to describe this in a landmark publication in 1965.5 After freezing plasma, the liquid could be removed and the precipitate stored in a plastic bag for up to one year, then thawed when needed for administration. The process was so simple and inexpensive that it was rapidly adopted around the world. As cryoprecipitate also contained other factors, it also provided a convenient treatment for people with von Willebrand’s disease.
References
- Patek AJ, Taylor FH. Hemophilia. II. Some properties of a substance obtained from normal human plasma effective in accelerating the coagulation of hemophilic blood. J Clin Invest 1937; 16: 113‒24.
- Pohle FJ, Taylor FH. The coagulation defect in hemophilia. The effect in hemophilia of intramuscular administration of a globulin substance derived from normal human plasma. J Clin Invest 1937; 16: 741‒7.
- Sibinga CT. Emile Rémigy and the discovery of anti-haemophilic activity in cryoprecipitate.Haemophilia 1996; 2: 56‒60.
- Pool JG, Hershgold EJ, Pappenhagen AR. High-potency antihaemophilic factor concentrate prepared from cryoglobulin precipitate. Nature 1964; 203: 312.
- Pool JG, Shannon AE. Production of high-potency concentrates of antihemophilic globulin in a closed-bag system. N Engl J Med 1965; 273: 1443‒7.
Further reading
Kasper CK. Judith Graham Pool and the discovery of cryoprecipitate. Haemophilia 2012; 18: 833‒5.
1967
Cryoprecipitate makes home care feasible read more
The development of plasma cryoprecipitate in 1964 and a system for large-scale production of a sterilised preparation in 1967 offered people with haemophilia and other bleeding disorders the prospect of routine factor replacement therapy. Whereas previously they had had to attend hospital for infusion of a large volume of plasma, they could now receive a high concentration of factor VIII or other clotting factors in a small volume of liquid. The use of frozen cryoprecipitate in the United States and of freeze-dried cryoprecipitate in Europe increased rapidly.
The new preparations were so easy and convenient to use that people with haemophilia were soon administering their treatment at home rather than attending a hospital clinic. Frozen cryoprecipitate required storage in a freezer at <20°C; the freeze-dried version could be kept in a fridge and reconstituted when needed, though the penalty for this convenience was a lower concentration of factor VIII.
By the early 1970s, home transfusions had become the preferred treatment for people with haemophilia. In 1972, one team from Chicago, US, described three years’ experience of home treatment for 32 people with haemophilia A.1 They encountered ‘minimal technical problems and no serious transfusion reactions’. Compared with hospital-based treatment, they recorded more bleeds; however, there were also fewer hospital admissions, fewer severe joint problems and fewer days off school or work. They also noted – and considered these of equal importance to the physical benefits of home treatment – a positive psychological effect on patients and their families, with increased self-reliance, better mobility and freedom from fear. They concluded: ’…until a prophylactic regimen is feasible, home transfusion is the treatment of choice for haemophilia.’
References
- Rabiner SF, Telfer MC. Home transfusion for patients with hemophilia A. N Engl J Med 1970; 283: 1011‒5.
1968
The first commercial Factor VIII concentrate manufactured read more
A 1968 brochure of blood products and accessories includes a monochrome advert with the picture of a 6ml syringe lying on its side, needle in place. It is promoting Hemofil, the first commercially available preparation of freeze-dried concentrated antihaemophilic factor (AHF, factor VIII).
The point of using the syringe was to emphasise that this was a product with a lower volume than the older but nonetheless very successful cryoprecipitate, and one that could be given by intravenous injection rather than infusion. (Of course, the syringe pictured represented the smallest volume of Hemofil available – the vials ranged in size from 4ml or 125 units to 30ml or 1,000 units.) Further, the vial was labelled with the concentration of AHF measured by assay. This was another advantage over cryoprecipitate, which contained a variable dose of AHF.
The AHF concentration in Hemofil was 100–400 times greater than in plasma1‒about six times greater than in cryoprecipitate ‒and it contained lower levels of extraneous proteins such as fibrinogen. It could be reconstituted and injected in a matter of minutes, whereas cryoprecipitate had to be thawed over 30 minutes and infused from a bag attached to a drip stand. It was undoubtedly a major improvement in convenience for people with haemophilia. Freeze-dried AHF concentrate was also available from blood transfusion services, but it contained about one-third of the AHF activity of Hemofil.2
Hemofil was manufactured by the Hyland Division of Travenol Laboratories, part of Baxter. However, it was prepared from pooled blood and, as was customary at the time, it was not treated to destroy possible viral contamination. It was withdrawn in 1975 after being associated with an outbreak of hepatitis B. Attempts to reintroduce a heat-treated formulation under the brand Hemofil T were discontinued when it was shown that this did not prevent transmission of hepatitis.
References
- Brinkhous KM, Shanbrom E, Roberts HR, et al. A new high-potency glycine-precipitated antihemophilic factor (AHF) concentrate: treatment of classical hemophilia and hemophilia with inhibitors. JAMA 1968; 205: 613‒7.
- Biggs R, Rizza CRC, Blackburn EK, et al. Factor VIII concentrates made in the United Kingdom and the treatment of haemophilia based on studies made during 1969‒72. Report of the Medical Research Council’s Blood Transfusion Research Committee Working Party on the cryoprecipitate method of preparing AHF concentrates. Br J Haematol 1974; 27: 391‒405.
1969
Armstrong and Aldrin walk on the moon read
more
Everybody knows the words Neil Armstrong spoke as he descended from the Eagle landing craft onto the surface of the moon on 20 July 20 1969: “That’s one small step for a man, one giant leap for mankind.” (Though when you listen to the recording it sounds more like “One small step for Man …” – try it: https://www.nasa.gov/wav/62284main_onesmall2.wav.)
Fewer people are aware that 12 astronauts have walked on the moon, the last being Gene Cernan and Harrison Schmitt in 1972. But it was Apollo 11 that blazed the trail to fame, only eight years after the first manned space flight by the Russian cosmonaut Yuri Gagarin. In 1961, President John F Kennedy had committed the United States to the space race, ostensibly in the pursuit of science, but in reality to demonstrate the superiority of American technology over its Cold War rival the USSR:
“I believe this nation should commit itself to achieving the goal before this decade is out, of landing a man on the moon and returning him safely to the earth. No single space project in this period will be more impressive to mankind, or more important for the long range exploration of space, and none will be so difficult or expensive to accomplish.”
Apollo 11 carried Neil Armstrong, Buzz Aldrin and Michael Collins to lunar orbit in the module Colombia. There, Armstrong and Aldrin climbed into the Eagle to make their descent. It was not a smooth journey – as shown in the 2018 movie First Man – and Armstrong navigated the craft manually to avoid surface debris, landing with only 30 seconds of fuel remaining.
Armstrong stepped out of the Eagle at 10.56 pm, followed shortly after by Aldrin. They spent about 2.5 hours on the surface, collecting samples and taking photographs, and planted the US flag, which appeared to fly proudly despite the lack of wind thanks to a telescopic rod that had not deployed fully. They then re-entered the Eagle and docked with Colombia for the return journey, splash landing off Hawaii on 24 July.
Further reading
NASA. A president issues NASA’s first historic challenge. Last updated 19 May 2011 (accessed April 2019).
NASA. July 20, 1969: one giant leap for mankind. 20 July 2017; last updated 31 January 2019 (accessed April 2019).
1971
A missing protein is identified as the cause of VWD read more
Erik Adolf von Willebrand described the bleeding disorder that was later to bear his name in 1926. He believed it was caused by dysfunction of platelets and the vascular wall. Only in 1971 was it discovered that the cause was an inherited deficiency of a protein that was associated with factor VIII and therefore christened factor VIII-related antigen.
Theodore Zimmerman and his colleagues from Cleveland, Ohio, applied the new science of immunoelectrophoresis to the problem.1 They developed an antiserum to antihaemophilic factor (AHF, now known as factor VIII) and used it to assay blood taken from people with ‘classic haemophilia’, von Willebrand disease (VWD), and controls who did not have a bleeding disorder. They found a protein antigenically similar to AHF in normal levels in controls and in people with haemophilia, but not in those with VWD. The test could therefore distinguish between haemophilia and VWD.
At the time, it was not known that the protein, then labelled AHF-like antigen (and today known as VWF:Ag), was von Willebrand factor (VWF), and that it occurred in blood as a complex with factor VIII as well as in platelets and the walls of blood vessels. VWF acts as a chaperone to factor VIII, preventing its rapid degradation. The factor VIII/VWF complex is split by the action of thrombin as part of the coagulation cascade.
Together with Italian haematologist Zaverio Ruggeri, Zimmerman later described ten forms of VWF with different physiological properties, providing the molecular basis for identifying subgroups of VWD.2 He died in 1988, aged 51.
References
- Zimmerman TS, Ratnoff OD, Powell AE. Immunologic differentiation of classic hemophilia (factor 8 deficiency) and von Willebrand’s disease, with observations on combined deficiencies of antihemophilic factor and proaccelerin (factor V) and on an acquired circulating anticoagulant against antihemophilic factor. J Clin Invest 1971; 50: 244‒54.
- Ruggeri ZM, Zimmerman TS. Variant von Willebrand’s disease: characterization of two subtypes by analysis of multimeric composition of factor VIII/von Willebrand factor in plasma and platelets. J Clin Invest 1980; 65: 1318‒25.
1973
Factor VIII separated from Von Willebrand factor read more
Von Willebrand disease is the most common inherited blood disorder, affecting about one per 1,000 people. The identification of von Willebrand factor (VWF) transformed scientific understanding of this disorder, paving the way for the development of specific treatments.
In 1973, Harvey J. Weiss and his team at Columbia University, New York, reported that the antibiotic ristocetin caused platelets from ‘normal’ blood to coagulate but did so weakly or not at all in blood from people with von Willebrand disease.1 They speculated that this was due to ‘a deficiency of a plasma factor necessary for platelet function’ that was somehow associated with factor VIII. In a second study, they concluded that what they were now calling von Willebrand factor was a protein associated with ‒ but distinct from ‒ factor VIII, and that a deficiency in this factor was the cause of von Willebrand disease.2
Subsequent research demonstrated the presence of VWF in the walls of blood vessels and platelets, and showed that it is released by the action of shear forces on the vascular wall. VWF was found to occur in blood as a chaperone bound to factor VIII, acting to prevent its rapid degradation in blood. In the coagulation cascade, the factor VIII/VWF complex is split by the action of thrombin; VWF then binds to platelets to promote aggregation. If there is a lack of VWF, or it does not function correctly, blood levels of factor VIII are reduced and platelets do not properly contribute to clot formation.
Changes in VWF are now the basis for the classification of von Willebrand disease into three main types. Type 1, which accounts for about 85% of cases, is due to a partial deficiency of VWF. Type 3, which is the rarest, affects about one per million people and is caused by an almost complete deficiency of VWF. Type 2 (of which there are four subtypes) is caused by abnormalities in function. It is believed that small variations in VWF function occur in the general population, giving rise to mild bleeding symptoms in some people who do not meet the diagnostic criteria for von Willebrand disease.
Depending on the severity of the bleeding risk, von Willebrand disease can be treated with desmopressin, a synthetic hormone that increases VWF levels, or with a concentrate of human plasma containing VWF and factor VIII. A recombinant form of human VWF was approved for marketing in Europe in 2018.
References
- Weiss HJ, Rogers J, Brand H. Defective ristocetin-induced platelet aggregation in von Willebrand’s disease and its correction by factor VIII. J Clin Invest 1973; 52: 2697‒707.
- Weiss HJ, Hoyer LW, Rickles FR, et al. Quantitative assay of a plasma factor deficient in von Willebrand’s disease that is necessary for platelet aggregation. Relationship to factor VIII procoagulant activity and antigen content. J Clin Invest 1973; 52: 2708‒16.
Further reading
Federici AB, Berntorp E, Lee CA. The 80th anniversary of von Willebrand’s disease: history, management and research. Haemophilia 2006; 12: 563–72.
Sharma R, Flood VH. Advances in the diagnosis and treatment of Von Willebrand disease. Blood 2017; 130: 2386–91.
1975
Monoclonal antibodies are developed read
more
Monoclonal antibodies have transformed modern medicine. They provide a way to target a specific molecule, such as a hormone, protein or its receptor, and block its actions. This means that a key component of a physiological pathway or a specific cell function that contributes to a disorder can be interrupted without directly affecting other parts of the body (though there are indirect effects). For example, tumour necrosis factor alpha is a major driver of inflammation in rheumatoid arthritis; when its actions are blocked by a monoclonal antibody, the severity of inflammation is greatly reduced and sometimes abolished. The symptoms of arthritis are greatly decreased and the progression of the disease is arrested. Monoclonal antibodies have proved so effective in the treatment of rheumatoid arthritis that they are now the medicines of choice for moderate or severe disease.1
The discovery of how to make monoclonal antibodies in quantities sufficient to be therapeutically useful was recognised by the 1984 Nobel prize in physiology or medicine.2 The prize was shared by Danish immunologist Niels Jerne (1911–1994), German biologist George Köhler (1946–1995) and Argentinian biochemist César Milstein (1927–2002) ‘for theories concerning the specificity in development and control of the immune system and the discovery of the principle for production of monoclonal antibodies.’
Jerne’s theories of systems biology led to the development of the clonal selection theory, which describes how the body’s B cells produce multiple identical copies of a single antibody (a ‘monoclonal’ antibody). He also developed an assay that enabled researchers to quantify the number of B cells producing antibodies.
B cells cannot be maintained for long in a laboratory environment, leading researchers to look for a more stable source of antibodies. Milstein and his colleagues developed a technique through which B cells could be fused with myeloma cells (bone marrow cancer cells) to create lines of ‘hybridoma’ cells that replicated repeatedly in the laboratory, producing large quantities of antibodies. However, the antibodies had no specific target.
Milstein described his work in a presentation at the Basel Institute, where George Köhler sat in the audience. Köhler subsequently joined Milstein’s team and worked out a way to target the antibodies. He activated B cells in mice by immunising them with an antigen, collected the B cells from the spleen and fused them with a myeloma cell line. Using Jerne’s assay technique, he then showed that the hybrid cells were producing identical antibodies targeted against the antigen. This is the basis of modern production techniques for monoclonal antibodies.
Monoclonal antibodies are used in many fields of medicine, with around half accounted for by cancer treatment, haematology, rheumatology, dermatology and gastroenterology. Potential therapeutic applications now being developed include their use in obesity, diabetes, coeliac disease, Alzheimer’s disease and bacterial infection. It is estimated that the global medicines market for these agents will be worth $130‒200 billion by 2022.
References
- Monaco C, Nanchahal J, Taylor P, Feldmann M. Anti-TNF therapy: past, present and future. Int Immunol 2015; 27: 55‒62.
- The Nobel prize in physiology or medicine 1984. NobelPrize.org. Nobel Media AB 2019 (accessed April 2019)
- Grilo AL, Mantalaris A. The increasingly human and profitable monoclonal antibody market. Trends Biotechnol 2019; 37: 9‒16.
1975
FEIBA (FVIII inhibitor bypassing activity) is launched read more
Regular administration of clotting factor (prophylaxis) is the treatment of choice to prevent bleeding and lower the risk of long-term joint damage in people with haemophilia. One of the most difficult challenges to the success of this approach is the development of antibodies – known as inhibitors ‒that neutralise the replacement clotting factor, greatly reducing its activity. This makes it difficult to control bleeds and increases the risk of complications. The existence of a factor VIII inhibitor was first reported in 1941.1
Inhibitors are most likely to develop against replacement factor VIII, as it is more antigenic than factor IX or von Willebrand factor (although they may rarely provoke inhibitors too). Risk factors include certain mutations of the haemophilia gene, Hispanic and African ethnicity, family history, and more intensive use of replacement factor.2Because the development of inhibitors is related to how much factor replacement has been used, it is largely a problem for people with severe haemophilia.
In the UK, 2.7% of the 8,000 or so people with haemophilia A (of all severities) who are registered with the National Haemophilia Database currently have inhibitors, and a further 4.7% have had them in the past.3 Of these, 0.4% and 0.7% of individuals respectively have levels high enough (at least 5 IU/dl) to have a significant impact on their treatment.
Low levels of inhibitors (5 IU/dl) can be overcome by increasing the dose of replacement factor. In people with severe haemophilia A, high inhibitor levels can be eradicated through immune tolerance therapy, which involves long-term treatment with very high doses of factor VIII to overwhelm the immune system. This typically takes 6–12 months, but may take up to three years. In the meantime, there are only two options for treating a bleed or preventing a bleed during surgery: recombinant factor VIIa or factor VIII inhibitor bypassing activity – also known as activated prothrombin complex concentrate, but more easily remembered by the brand name FEIBA.
The possibility that prothrombin concentrate prepared from human plasma contained something that could bypass factor VIII was first raised in 1969,4 and confirmed in 1972 and 1974.5,6 FEIBA, Europe’s first commercial preparation of activated prothrombin complex – which also contains factor Xa and prothrombin ‒was introduced in 1975 by the Austrian manufacturer Immuno. It promotes haemostasis when factor VIII function is inhibited by contributing to the coagulation cascade immediately after the step where factor VIII acts, increasing thrombin generation. It also bypasses the step at which factor IX is involved, and is therefore effective in people with haemophilia B and inhibitors.
References
- Lawrence JS, Johnson JB. The presence of a circulating anti-coagulant in a male member of a hemophiliac family. Trans Am Clin Climatol Assoc 1941; 57: 223–31.
- Collins PW, Chalmers E, Hart DP, et al. Diagnosis and treatment of factor VIII and IX inhibitors in congenital haemophilia: (4th edition). UK Haemophilia Centre Doctors Organization. Br J Haematol 2013; 160: 153–70.
- United Kingdom Haemophilia Centre Doctors’ Organisation and National Haemophilia Database. UKHCDO annual report 2018 including bleeding disorder statistics for 2017/2018. October 2018 (accessed April 2019).
- Breen FA Jr, Tullis JL. Prothrombin concentrates in treatment of Christmas disease and allied disorders. JAMA 1969; 208: 1848‒52.
- Fekete LF, Holst SL, Peetoom F, et al. “Auto” factor IX concentrate: a new therapeutic approach to treatment of hemophilia A patients with inhibitors. XIV Congress of the International Society of Haematology. Sao Paulo, Brazil. 1972. (Abstract 295).
- Kurczynski EM, Penner JA. Activated prothrombin concentrate for patients with factor VIII inhibitors. N Engl J Med 1974; 291: 164‒7.
1977
Desmopressin, a synthetic version of vasopressin, is developed read more
In 1974, a team from the Regional Blood Transfusion Centre in Edinburgh’s Royal Infirmary published the first evidence that desmopressin, a synthetic analogue of the hormone vasopressin, increased blood levels of factor VIII and plasminogen activator in healthy volunteers.1 There was a clear relationship between the dose of desmopressin and the level of factor VIII, and the response was ‘sustained and without side effects’.
It was not until 1977 that a study testing this observation in clinical practice was published.2 A team of haematologists from Milan, Italy, administered infusions of desmopressin to patients with mild or moderate haemophilia or von Willebrand disease (VWD) shortly before and soon after dental extraction surgery. They observed a two- to three-fold increase in factor VIII activity in the blood, which prevented bleeding in two patients and limited bleeding to oozing from the gums in two others (for which they needed clotting factor replacement). A higher dose increased factor VIII activity up to normal levels in another six people with mild haemophilia and two with VWD.
That study was timely. In the late 1970s, clotting factor was in short supply and what little was available was reserved largely for on-demand treatment. Furthermore, blood products had become contaminated with hepatitis virus and their use could not be justified for people with mild bleeding disorders whose greatest risk was from trauma and surgery rather than spontaneous bleeds. Desmopressin was a safe, predictable option. Its widespread use in Italy is believed to have been a factor in the low prevalence of HIV infection among people with mild haemophilia A compared with those with mild haemophilia B, who required treatment with plasma products because desmopressin does not increase levels of factor IX.3
However, early studies did not detect the adverse effects of desmopressin that are now recognised. These include flushing, hypotension and fluid retention, which requires restriction of liquids intake for 24 hours after a dose. The response is variable and a test dose is administered to determine how useful it will be in individual cases.4
Desmopressin increases levs of factor VIII and von Willebrand factor levels by stimulating their release from the endothelial cells of blood vessels.4 It is effective in people with VWD and mild or moderate haemophilia, but not in those with severe haemophilia or type 3 VWD as they have little or no stores of factor VIII or von Willebrand factor. It has many advantages: it is less expensive than treatment with a clotting factor and avoids the risk of developing inhibitors; it appears to be safe during pregnancy; and it is administered subcutaneously (or intranasally), offering greater convenience. It is therefore preferred to blood products for treating and preventing bleeds in people with mild or moderate haemophilia A or VWD, and for symptomatic carriers of haemophilia A.5
References
- Cash JD, Gader AM, da Costa J. Proceedings: The release of plasminogen activator and factor VIII to lysine vasopressin, arginine vasopressin, I-desamino-8-d-arginine vasopressin, angiotensin and oxytocin in man. Br J Haematol 1974; 27: 363‒4.
- Mannucci PM, Ruggeri ZM, Pareti FI, Capitanio A. 1-Deamino-8-d-arginine vasopressin: a new pharmacological approach to the management of haemophilia and von Willebrands’ diseases. Lancet 1977; 1: 869‒72.
- Mannucci PM. Use of desmopressin in the treatment of hemophilia A: towards a golden jubilee. Haematologica 2018; 103: 379‒81.
- Laffan MA, Lester W, O’Donnell JS, et al. The diagnosis and management of von Willebrand disease: a United Kingdom Haemophilia Centre Doctors Organization guideline approved by the British Committee for Standards in Haematology. Br J Haematol 2014; 167: 453–65.
- World Federation of Hemophilia. Guidelines for the management of hemophilia. 2nd ed. 2012 (accessed April 2019).
1982
The Haemophilia Nurse Association holds its first meeting read more
We hear a lot about disruptive therapies these days – innovations so radical that they have the potential to bring about a drastic change in treatment and also in the way care is organised and funded. Extended half-life clotting factors and gene therapy are two recent examples of disruptive therapies. But it is not widely known that the Haemophilia Nurses’ Association (HNA) was founded in response to a disruptive therapy that fulfilled its potential to fundamentally transform haemophilia care.
Clotting factor concentrate, introduced in the 1960s, was a disruptive therapy. It made life much easier for people with haemophilia, as bleeds could be treated with a low volume infusion rather than large volumes of plasma. Patients would normally visit hospital to have the drip set up and venepuncture carried out. However, it soon became evident that this was a simple procedure that could be done at home by the patients themselves, with training and support from the clinic. By 1970, leading centres in the United States were strongly advocating this approach.1
In the UK, people were still attending hospital to get treatment for bleeds, usually receiving their infusion of concentrate as an outpatient. At the time, the nurse’s role did not even include taking bloods or setting up intravenous infusions, but forward-thinking individuals were starting to change practice. Maureen Fearns, a haemophilia sister in Newcastle upon Tyne, was one such pioneer. The Newcastle haemophilia centre was progressive: the director had recently established a comprehensive care team to deliver holistic care to patients and their families. Maureen was keen to learn about clinical practice in other centres: “Initially, I sent out a one page newsletter to nurses working with haemophilia patients at other centres, asking them what they were doing and how they were doing it, and whether they’d be interested in sharing information – and we just took it from there.”2
That letter led to the first meeting of haemophilia nurses in London in 1976, at which Maureen described her experience of setting up a home treatment service for adults and children with severe haemophilia. That event and the meetings that followed were such a success that, in 1981, she and Patricia Turk from the Treloar School and College for young people with disabilities formally established the HNA. Its first meeting was held in Newcastle’s Airport Hotel over two days in September 1982, with home therapy training as the first presentation.
From its inception, the HNA aimed to improve patient care through nurse education and training, promoting communication and support within the specialty, building links with professional societies and other disciplines, and developing professional standards. The experience of caring for patients with HIV and their families during the 1980s, and again for those affected by hepatitis in the 1990s, reinforced the bonds within the HNA, demonstrating that its network, meetings and peer support were highly valued by its members. The role of haemophilia nurses has since greatly expanded and now includes every aspect of the organisation and delivery of care, with specialist haemophilia nursing posts now formally recognised.
Today the HNA has around 120 members. In partnership with Haemnet, it provides courses on clinical practice and leadership and is developing a new competency framework. It is actively strengthening links with other healthcare professionals, such as physiotherapists, and reaching out to haemophilia nurses throughout Europe. Maureen was invited to address the HNA Annual Meeting in Birmingham on 30 March 2019, where she recounted how the experiences of its members over more than three decades had made the Association even more relevant to nursing practice, providing a stronger voice for nurses and patients alike.
References
- Rabiner SF, Telfer MC. Home transfusion for patients with hemophilia A. N Engl J Med 1970; 283: 1011‒5.
- Bryan J. How nurses built a specialty from scratch. J Haem Pract 2014; 1: 28‒31. doi: 10.17225/jhp.00009.
1982
The first cases of AIDS reported in people with haemophilia read more
In July 1982, the United States Centers for Disease Control (CDC) reported that three people with haemophilia A had developed a rare form of pneumonia due to the fungus Pneumocystis carinii(now known as Pneumocystis jirovecii). All died.1,2
The organism responsible typically causes infection in people whose immune system is compromised, such as in the severe immune deficiency syndrome in homosexual men and intravenous drug users that had been reported in New York and California in 1981. ‘The clinical and immunologic features these three patients share are strikingly similar’ to the cases reported in people with immune deficiency, the CDC noted, adding that the cause was unknown but ‘the occurrence among the three hemophiliac cases suggests the possible transmission of an agent through blood products.’
By December 1982, it was clear that the three men with haemophilia had what was now known as Acquired Immune Deficiency Syndrome (AIDS). Five similar cases had also come to light: three were children and two adults were already dead. The only risk factor common to them all was treatment with factor VIII concentrate or other blood products.
Lymphadenopathy-associated virus, later renamed human immunodeficiency virus (HIV), was eventually confirmed as the cause of AIDS in 1983.3Isolated cases had been reported for many years without understanding the role of the virus, but since 1980 its increasing frequency in high-risk groups had attracted medical attention.
HIV had entered the blood supply in the United States because donations were accepted from people in high-risk groups. Public health authorities warned people who used blood products of the risk, but encountered resistance to their proposal that blood should not be accepted from high-risk individuals – including the already stigmatised gay community, which accounted for a high proportion of donations.4Scepticism among representatives of the haemophilia community, the US medicines regulator and the blood industry also delayed progress. Blood banks refused to provide donors’ details, obstructing attempts to connect transfusion and transmission. However, in December 1982, the CDC proved the link when it reported that a 20-month‐old infant had developed AIDS after receiving a transfusion of platelets derived from the blood of a man later found to have AIDS.
Doubt and denial persisted but, in March 1983, the CDC recommended that blood donations should not be accepted from high-risk groups.5By this time, over 1,200 cases of AIDS had been notified and 450 people had died; 11 cases were known among people with haemophilia, which increased to 26 by August. Attempts were already underway to develop methods to destroy HIV in blood products, and by October 1984 it could be shown that heat treatment removed the virus completely. The new year saw an almost complete switch to heat-treated blood products.
Between 1981 and 1984, more than half of all people with haemophilia in the United States had been infected with HIV. By 1998, 2,254 people with haemophilia had died of AIDS-related complications.6
During the 1980s, the UK was not self-sufficient in blood products and imported supplies from the United States. Between 1979 and 1986, 1,227 people with haemophilia were infected with HIV.7Between 1985 and 1992, 403 HIV-positive people died ‒the expected number had been 60. It was estimated that 85% of the excess deaths were due directly to the complications of AIDS. In 2017/18, one person registered on the National Haemophilia Database died of AIDs.
References
- Centers for Disease Control (CDC). Pneumocystis carinii pneumonia among persons with hemophilia A. MMWR Morb Mortal Wkly Rep 1982; 31: 365‒7.
- Centers for Disease Control (CDC). Update on acquired immune deficiency syndrome (AIDS) among patients with hemophilia A. MMWR Morb Mortal Wkly Rep 1982; 31: 644‒6, 652.
- Gallo RC, Montagnier L. The discovery of HIV as the cause of AIDS. N Engl J Med 2003; 349: 2283‒5.
- Evatt BL. The tragic history of AIDS in the hemophilia population, 1982-1984. J Thromb Haemost 2006; 4: 2295‒301.
- Centers for Disease Control (CDC). Prevention of acquired immune deficiency syndrome (AIDS): report of inter-agency recommendations. MMWR Morb Mortal Wkly Rep 1983; 32: 101‒3.
- Chorba TL, Holman RC, Clarke MJ, Evatt BL. Effects of HIV infection on age and cause of death for persons with hemophilia A in the United States. Am J Hematol 2001; 66: 229‒40.
- Darby SC, Ewart DW, Giangrande PL, et al. Mortality before and after HIV infection in the complete UK population of haemophiliacs. UK Haemophilia Centre Directors’ Organisation. Nature1995; 377: 79‒82.
- United Kingdom Haemophilia Centres Doctors Organisation.National Haemophilia Database. UKHCDO annual report 2018 including bleeding disorder statistics for 2017/2018. October 2018 (accessed April 2019).
1982
The factor IX gene is cloned read more
In September 1982, a research team from Oxford University reported that they had partially cloned the human gene for factor IX.1 Concluding the densely-packed description of their work, the authors commented, ‘It is hoped that the genomic clone reported here will be similarly useful for the diagnosis and understanding of the mutations in patients with haemophilia B, as well as of heterozygote mothers who are carriers of the factor IX mutation.’
Within two months, researchers from the University of Washington, Seattle, described the complete cloning of the factor IX gene, known as pHfIX1.2 It is sobering to see the gene’s sequence of amino acids typed out on a single page in this short scientific paper (available free online) and realise that this was the discovery that transformed the lives of people with haemophilia B.
Cloning the factor IX gene opened the way for the development of recombinant factor IX for replacement therapy, giving people with haemophilia B a safe source of replacement clotting factor. It also marked the point at which gene therapy for haemophilia B became a reality.
What is less well known is that cloning the factor IX gene also led to groundbreaking experiments with cloned sheep. Following the successful cloning of Dolly the sheep in 1996, researchers at Edinburgh’s Roslin Institute cloned two more: Molly and Polly. Like Dolly, they were created from a single cell of an adult sheep, but they were also transgenic – that is, their cells had been altered to express a non-native gene that would cause human factor IX to be expressed in their milk. This was the first experiment to show that transgenic animals could be used as a source of complex proteins to treat human disorders. The technique, known as pharming, is now used to manufacture recombinant human antithrombin (ATryn) from the milk of transgenic goats and the recombinant C1-inhibitor conestat alfa (Ruconest) from the milk of transgenic rabbits. Pig milk appears to offer the best prospect of transgenic production of recombinant clotting factors for therapeutic use.
References
- Choo KH, Gould KG, Rees DJ, Brownlee GG. Molecular cloning of the gene for human anti-haemophilic factor IX. Nature 1982; 299: 178‒80.
- Kurachi K, Davie EW. Isolation and characterization of a cDNA coding for human factor IX. Proc Natl Acad Sci USA 1982; 79: 6461‒4.
- Van Cott KE, Monahan PE, Nichols TC, Velander WH. Haemophilic factors produced by transgenic livestock: abundance that can enable alternative therapies worldwide. Haemophilia 2004; 10 Suppl 4: 70‒6.
1983
The first commercially available mobile phone read more
The early 1980s were a time of unusual technological innovation. The compact disc, the Apple Macintosh, the first Windows operating system and the Smartmodem (the forerunner of today’s link between home and the internet) all first saw the light of day in these years. But one gadget launched on an eager public in 1983 was to have profound effects on almost everyone: the mobile phone.
The Motorola DynaTAC 8000X offered 30 minutes of talk-time, storage for 30 phone numbers and six hours’ standby. It took ten hours to charge and cost over £2,600. In hindsight, it’s hard to see how it could ever be cool to walk the streets pressing a device the size and weight of a brick firmly to the head, but it soon was. Yuppies, those archetypal 1980s wide boys, were probably the only people able – or prepared – to pay the exorbitant price for a phone with limited storage, range and battery life. In an era synonymous with material acquisition and flagrant displays of wealth, the mobile phone had found its first champions.
The first public mobile phone call is credited to a Motorola executive named Martin Cooper. On 3 April 1973, he stood in the streets of New York and, using a phone weighing 1.1 kg, he called Bell Labs, Motorola’s main competitor. He made the call to Bell on its landline because it had just lost the race to launch the first mobile phone. Cooper is reputed to have joked that the talk time would have to be short because no one had the strength to hold the phone for any longer.
The first public mobile phone call in the UK was made on 1 January 1985 by the comedian Ernie Wise. Using the Transportable Vodafone VT1, weighing in at 5kg, he called Vodafone from London’s Docklands‒which, fittingly enough, was undergoing massive regeneration. By the end of 1985, Vodafone had sold over 12,000 of these devices.
The next giant leap forward came in 2007, with the launch of Apple’s iPhone. Within six months, iPhone sales reach 3.7 million; in 2018, Apple sold 2.2 billion iPhone XRs. It is now estimated there are more than 7 billion mobile phone numbers in use, with by far the most in China (1.4 billion) and India (1.2 billion). The UK ranks seventeenth in the world, with about 83 million.
Further reading
History of mobile phones. Wikipedia (accessed April 2019).
Greene B. 38 years ago he made the first cell phone call. CNN. 3 April 2011 (accessed April 2019).
Mobile phone history (accessed April 2019).
Costello S. How many iPhones have been sold worldwide? Lifewire. Updated 17 January 2019 (accessed April 2019).
1984
The Factor VIII gene is sequenced read
more
The human factor VIII gene was fully cloned in 1984 by researchers working at US biotech startup Genentech, based in San Francisco. This was the step that opened the way for production of recombinant human factor VIII, providing a safer alternative to the plasma-derived products then in use. Jane Gitschier, one of the team leaders, has given a colourful and very detailed account of the planning and research that led to the discovery.1
In the early 1980s, Genentech was an unlikely setting for such prestigious research. Gitschier recalls that the company ‘operated like a boy’s locker room, and the place reeked of testosterone. No prank was too outrageous, no poker bet was too high, and no woman was part of the inner circle. The head of organic chemistry came out of the building one day to find his car had been painted pink.’ Such boisterousness does not seem to have interfered with the serious business of research.
Despite Gitschier’s clear account, the explanation of how the Genentech team overcame the many practical and theoretical problems in their path requires a good understanding of gene biochemistry: the work was complex, technical and involved several teams investigating complementary lines of research, including researchers from the Haemophilia Centre at London’s Royal Free Hospital. The project started in 1981; as the end of 1983 approached, Genentech had sequenced almost all of the factor VIII gene.
But Genentech had a rival. Genetics Institute (GI), based in Washington, had been studying porcine factor VIII. In December 1983, the company shocked Genentech when it announced to the press that it had cloned the human factor VIII gene. It turned out that GI had actually only partially cloned the gene, though this did not deter the company from filing a patent based on what it knew of the sequence.
Genentech fully achieved its objective in April 1984, sequencing the entire gene and also expressing the gene from recombinant DNA. A patent was filed on the same day that this achievement was announced to the world.
The research was published in three papers in a single issue of the journal Nature,2-4 together with a paper from GI describing their work.5 GI marketed its recombinant factor VIII (Recombinate) in 1992; Genentech launched Kogenate in 1993. The two companies entered what was to become a patent dispute lasting five years.6 This was eventually settled in Genentech’s favour and the two companies agreed a cross-licence arrangement. GI was later taken over by Wyeth, which introduced a modified recombinant factor VIII under the brand ReFacto in 2000.
References
- Gitschier J. Remembrances of factor VIII. Part 1: the race to the gene. J Thromb Haemost 2004; 2: 383‒7.
- Gitschier J, Wood WI, Goralka TM, et al. Characterization of the human factor VIII gene. Nature 1984; 312: 326‒30.
- Wood WI, Capon DJ, Simonsen CC, et al. Expression of active human factor VIII from recombinant DNA clones. Nature 1984; 312: 330‒7.
- Vehar GA, Keyt B, Eaton D, et al. Structure of human factor VIII. Nature 1984; 312: 337‒42.
- Toole JJ, Knopf JL, Wozney JM, et al. Molecular cloning of a cDNA encoding human antihaemophilic factor. Nature 1984; 312: 342‒7.
- Genentech. Genentech receives favorable ruling on process patent application for recombinant human factor VIII:C. 21 August 1995 (accessed April 2019).
1988
The first use of recombinant Factor VIII in Europe read more
Following the successful cloning of the factor VIII gene in 1984, the first recombinant factor VIII to be marketed in Europe was Kogenate. The recombinant protein was produced in hamster kidney cells and there were concerns that residual animal antigens might provoke an immune response ‒in particular, the neutralising antibodies known as inhibitors. On the other hand, manufacturing clotting factors by recombinant technology avoided the risk of viral contamination of plasma products (this was by now more theoretical than real, but memories of HIV and hepatitis transmission were fresh).
The first clinical trial of Kogenate began in Europe and the United States in 1988.1 It involved three phases: a comparison of the pharmacokinetics of recombinant factor VIII with a plasma-derived product in 17 people; a study of the feasibility of home treatment involving 26 people; and the efficacy and safety of the recombinant factor as on-demand treatment for serious bleeds and to provide cover during surgery in 76 people (some of whom had participated in the preliminary studies). Twenty infants and children who had not previously received a clotting factor were treated with the recombinant product on demand or as prophylaxis.
The pharmacokinetics of recombinant factor VIII were found to be similar to those of the plasma product. Home treatment for almost two years showed that average infusion frequency was once per week and the average annual bleed rate was 24.4 (equivalent to one bleed every 14–15 days). Haemostasis was rated ‘excellent’ as cover for surgery. Inhibitors were found in eight patients; they predated recombinant therapy in one but were high-level (and therefore likely to reduce the effectiveness of replacement factor VIII) in two patients.
The Kogenate trial suggested that recombinant factor VIII was about as safe and effective as the plasma-derived products it might replace, though the authors acknowledged that questions remained about inhibitor risk. This debate rumbled on until, in 2016, the SIPPET study showed that recombinant products were associated with a 69% greater risk of causing high-level inhibitors than plasma products in previously untreated patients.2 Why this should be so remains unclear, but the conclusion has withstood criticism about its design and possible confounding factors.3
Reference
- Schwartz RS, Abildgaard CF, Aledort LM, et al.Human recombinant DNA-derived antihemophilic factor (factor VIII) in the treatment of hemophilia A. Recombinant Factor VIII Study Group. N Engl J Med 1990; 323: 1800‒5.
- Peyvandi F, Mannucci PM, Garagiola I, et al. A randomized trial of factor VIII and neutralizing antibodies in hemophilia A. N Engl J Med 2016; 374: 2054‒64.
- Peyvandi F, Mannucci PM, Palla R, Rosendaal FR. SIPPET: methodology, analysis and generalizability. Haemophilia 2017; 23: 353‒61.
1990
UK Haemophilia Chartered Physiotherapists Association (HCPA) established read
more
The Royal Victoria Hospital (RVI) in Newcastle upon Tyne has made (at least) two valuable contributions to the development of professional care in NHS haemophilia services. The first was the founding of the Haemophilia Nurses’ Association (HNA) by Maureen Fearns in 1982. The second was the Haemophilia Chartered Physiotherapists Association (HCPA), which held its first meeting in 1990 (albeit in London), organised by Brenda Buzzard, a physiotherapist at the RVI, and her colleagues Karen Beeton, Ruth Bolton, Fiona Hall and Fiona McChesney.
The original aim of the HCPA was to provide a forum for communication via meetings and a newsletter ‒ an ambition that was soon met through a series of meetings in haemophilia centres throughout the UK. It was clear that there was great enthusiasm within the profession for improving the quality of care by developing practice standards and collaborating with agencies such as the World Federation for Hemophilia (WFH).
With support from Bayer, HCPA was able to develop its own continuing professional development (CPD) training and liaise more widely in the international forums. In 2007, an annual meeting was established to promote research, facilitate networking and provide peer support. Since then, the Association has established standards of care for child and adult haemophilia care. It has formed a clinical studies group to provide support and mentorship for researchers, and has strengthened relationships with the HNA. There is also a web-based peer-support forum on Haemnet (www.haemnet.com), where members can meet and share problems and issues. HCPA represents physiotherapists at the NHS England Clinical Reference Group by helping to develop the service specification for England, and is a member of the outcomes and musculoskeletal working parties of the UK Haemophilia Doctors’ Organisation. Further afield, HCPA members are represented on the physiotherapy groups of both the European Association for Haemophilia and Allied Bleedings Disorders (EAHAD) and the WFH.
The HCPA has, in a relatively short period, established the profession’s credentials and created the foundation on which physiotherapists working with people with haemophilia can further improve care by promoting research and good clinical practice. It provides professional leadership and a forum for exchanging ideas within the profession and with colleagues working in haemophilia care.
Further reading
Bladen M, Stephenson D, McLaughlin P. UK physiotherapy and haemophilia: A future strategy built on past success. J Haem Pract 2016; 3(2): 15‒20. doi: 10.17225/jhp00076.
1990
Tim Berners-Lee invents the World Wide Web read more
Few innovations have brought such change for as many people as the world wide web. Thanks to this creation, which UK physicist Sir Tim Berners-Lee made freely available for the good of all, half the world is now online and able to communicate, with all the educational, social and commercial benefits that come with it. In only 30 years, it has become easier to have a face-to-face conversation with someone on the other side of the world than to send a postcard. Those of us who find it difficult to understand the technology underpinning this revolution could reasonably call it ‘magic’.
Every year, Tim Berners-Lee publishes a letter in which he comments on the impact of the world wide web (https://webfoundation.org). In 2019, the web’s thirtieth anniversary, he was hopeful but concerned.
First, he makes the often-overlooked point that every step forward for the web widens the gap between those who have access and those who do not. And with every step forward, that gap gets harder to cross. He writes:
‘It is more urgent than ever to ensure the other half are not left behind offline, and that everyone contributes to a web that drives equality, opportunity and creativity.’
Mostly, however, he recognises the good and bad uses to which the world has put his creation:
‘…while the web has created opportunity, given marginalised groups a voice, and made our daily lives easier, it has also created opportunity for scammers, given a voice to those who spread hatred, and made all kinds of crime easier to commit.
‘Against the backdrop of news stories about how the web is misused, it’s understandable that many people feel afraid and unsure if the web is really a force for good. But given how much the web has changed in the past 30 years, it would be defeatist and unimaginative to assume that the web as we know it can’t be changed for the better in the next 30. If we give up on building a better web now, then the web will not have failed us. We will have failed the web.’
Berners-Lee has proposed a new contract for the web, calling on citizens, corporations and governments to act. Governments must introduce legislation fit to protect people in the digital age; corporations should stop exploiting their clients and customers for their own interests; citizens must hold governments and corporations accountable. It’s a call that shows his faith in humanity is undimmed – the same faith that led him to give away one of the greatest inventions of all time. He concludes that the fight for the web is ‘one of the most important causes of our time’:
‘It’s our journey from digital adolescence to a more mature, responsible and inclusive future.
‘The web is for everyone and collectively we hold the power to change it. It won’t be easy. But if we dream a little and work a lot, we can get the web we want.’
Further reading
Berners-Lee T. 30 years on, what’s next #ForTheWeb? World Wide Web Foundation. 12 March 2019 (accessed April 2019).
For The Web (accessed April 2019).
Hern A. Tim Berners-Lee on 30 years of the world wide web: ‘We can get the web we want’. The Guardian. 12 March 2019 (accessed April 2019).
1991
UKHCDO is registered as a charity read
more
The UK Haemophilia Centres Doctors’ Organisation (UKHCDO) was founded in 1968 to ‘improve haemophilia care, research into bleeding disorders, their treatment, epidemiology and complications, and to facilitate healthcare planning.’ Ten years later it had established the UK’s registry of people with bleeding disorders, the National Haemophilia Database (NHD). It became a registered charity in 1991.
Despite its name, UKHCDO is concerned with all bleeding disorders. Its objectives are defined as:
- to preserve, protect and relieve persons suffering from haemophilia and other inherited bleeding disorders;
- to advance the education of the medical profession, the nursing profession, professions allied to medicine and the general public in the knowledge of haemophilia and other inherited bleeding disorders and their treatment;
- to promote or assist in the promotion of audit and research into the causes, prevention, alleviation and management of haemophilia and other inherited bleeding disorders and to disseminate the useful results of such research.
The NHD (http://www.ukhcdo.org/nhd) is a treasure trove of information about people with haemophilia in the UK and their care. The UKHCDO is required by the Department of Health to collect data from haemophilia centres to help to plan haemophilia care and determine spending commitments, and to facilitate research. The NHD is mainly funded by the NHS, with additional support from the pharmaceutical industry for research and information for medicines regulators. Annual reports from the NHD are publicly available at http://www.ukhcdo.org/home-2/annual-reports.
The UKHCDO has 11 working parties and task forces, focusing on many aspects of haemophilia and its management and publishing authoritative management guidelines. It also liaises with other professional groups, including the Haemophilia Nurses’ Association, the Haemophilia Chartered Physiotherapists’ Association, the Haemophilia Society and the Clinical Molecular Genetics Society. It maintains a register of UK haemophilia centres and operates the Haemtrack system, through which people with haemophilia monitor their treatment and coordinate with their treatment centres.
1992
Recombinant Factor VIII concentrates licensed read more
The biotech company Genetic Industries (GI) was – just – beaten to the line by Genentech for the first sequencing of the entire factor VIII gene. However, it was the first to bring to market a recombinant human factor VIII. Recombinate was introduced in the United States in 1992, where it is still available.
Recombinant factor VIII marked the final move away from using human blood as a source of clotting factor ‒a necessary change following the contamination of donated blood supplies by HIV and hepatitis virus, and a shortage of blood from safe sources. Newer formulations of clotting factors had been heat-treated and cleared by ultrafiltration, so the risk of viral transmission was negligible by this time. However, the risk of previously unrecognised pathogens could not be ruled out – the UK outbreak of bovine spongiform encephalopathy had raised concerns that prions might be potential contaminants. Confidence in human blood sources therefore remained low; recombinant products offered safety and greater certainty about supply.
The early recombinant clotting factors were not entirely free of human blood products. Recombinate (antihaemophilic factor [recombinant], later available in Europe as Helixate), the only example of a ‘first generation’ recombinant factor VIII, contains human albumin as a stabiliser and, unlike later products, was not treated to remove viruses. Second generation agents, such as Kogenate (octocog alfa) and ReFacto (moroctocog alfa) have a different stabiliser. but may contain traces of human albumin from the cell culture medium in which the compounds are synthesised. The third generation recombinant factor Advate (octocog alfa), licensed in Europe in 2004, is a modified formulation of Recombinate from which all animal and human products have been removed during fermentation and manufacture.
1994
WFH introduces its twinning programs read
more
With the advent of gene therapy, it is remarkable that it is now possible to talk – however tentatively – about a cure for haemophilia. But this further highlights the acknowledged disparities in the care for people with bleeding disorders available in wealthy developed countries and economically developing countries. The World Federation of Hemophilia (WFH) has been working for over 50 years to close this therapeutic gap, and one of its most effective approaches is the twinning programme.
The idea of a twinning programme was first proposed by Guglielmo Mariani, now a professor of haematology at the Università LUdeS, Lugano, Switzerland. His original plan has changed little: twinning is a formal, two-way collaboration or partnership between emerging and established treatment centres or organisations, and builds personal and organisational relationships that encourage the sharing of knowledge and experience to the benefit of both partners. WFH is adamant that this is not a one-way process by which the wealthier twin gives and the other takes, noting that relationships in which one centre views itself as ‘the expert’ are at risk of being unbalanced. Over the past 20 years, the WFH Twinning Programme has established 215 partnerships in 113 countries.
Types of twinning activities include exchange visits, training, sharing expertise on complex cases, swapping information resources, supplies of equipment, reagents and factor concentrates, special projects such as advocacy and outreach, and research and networking. Both twins benefit from capacity building, sharing best practice and collaborative working.
In 2016, WFH launched a new twinning initiative to help improve and strengthen youth groups in developing partner countries, with the specific aim of fostering leadership.
The twinning programme is funded exclusively by Pfizer.
Further reading
Giangrande PL, Mariani G, Black C. The WFH Haemophilia Centre Twinning Programme: ten years of growth, 1993-2003. World Federation of Hemophilia Occasional Papers: no. 4, 2003 (accessed April 2019).
Mariani G. The Haemophilia Centre Twinning Programme. Haemophilia 1996; 2: 125‒7.
World Federation of Hemophilia. Improving care beyond our borders: a twinning guide for hemophilia treatment centres. 2002 (accessed April 2019).
World Federation of Hemophilia. Our work: Twinning program. Updated March 2017 (accessed April 2019).
1997
Recombinant Factor IX products available read more
On 11 February 1997, US medicines regulator the Food and Drug Administration notified biotech company Genetics Institute that it was now authorised to ‘manufacture and ship for sale, barter or exchange in interstate and foreign commerce Coagulation Factor IX (Recombinant).’1
The protein, now known as nonacog alfa, was synthesised using a stable line of Chinese hamster ovary cells and was free of animal and human products. Factor IX was marketed under the brand BeneFIX. In Europe, BeneFIX was licensed on 27 August 1997. Children and adults with haemophilia B now had their first recombinant clotting factor.
Prior to 1997, factor IX products were derived from human plasma. Initially, these were extracts that contained only low levels of factor IX, but also factors II and VII; they were therefore known as prothrombin complex concentrates. These crude products were associated with a potential risk of thrombosis and were replaced by high purity alternatives that had undergone heat treatment and ultrafiltration to remove possible viral contamination. But as recently as 1996, US patients had been warned of a possible risk of hepatitis A transmission associated with high purity products.2
In the UK today, 69% of factor IX products prescribed for people with haemophilia B are high purity recombinant proteins.3 BeneFIX still dominates the market, accounting for 98% of recombinant factor IX products, but its use is now declining with the advent of extended half-life factors. This next generation of recombinant proteins has been modified to reduce the rate at which factor IX is eliminated from the body. Introduced in 2016, they made up 23% of all factor IX products prescribed in 2017/18.
References
- US Food and Drug Administration. February 11, 1997 approval letter – Benefix (accessed April 2019).
- Centers for Disease Control and Prevention (CDC). Hepatitis A among persons with hemophilia who received clotting factor concentrate ‒United States, September-December 1995. MMWR Morb Mortal Wkly Rep 1996; 45: 29‒32.
- United Kingdom Haemophilia Centres Doctors’ Organisation and National Haemophilia Database. UKHCDO annual report 2018 including bleeding disorder statistics for 2017/18. October 2018 (accessed April 2019).
1998
The first gene therapy trials for haemophilia begin read more
The 1990s were a time of feverish activity as researchers struggled to develop a gene therapy for haemophilia. The concept had been demonstrated in experimental models, but proof was needed that it would work in humans. The first clinical trial to attempt that began in 1998.1 By current standards the results were disappointing.
The trial was a Phase I study; that is, it was designed to assess dosage and side effects. A team from Boston, Massachusetts, working with biotech company Transkaryotic Therapies, took fibroblasts from the skin of six people with severe haemophilia A, transfected them with parts of the gene for factor VIII, cultured the cells and reinjected them into the peritoneum.
After one year, no serious adverse effects were detected. Factor VIII activity increased in four participants, coinciding with a reduction in bleeding frequency and use of clotting factor. However, the highest factor VIII activity was only 4%, and that was recorded in only one person after three months; after one year, factor VIII activity had returned to baseline levels in all participants.
Transkaryotic Therapies was not the only player in an area of research that offered huge prizes for success.2It was clear that gene therapy could work if only the person’s cells could be persuaded to take up the gene in a way that ensured it would survive and produce factor VIII. Researchers turned to viruses to use as vectors deliver the gene. Chiron Inc tried intravenous administration using a mouse leukaemia virus; it failed. GenStar Therapeutics Inc tried using an adenovirus, but this provoked an immune reaction that destroyed the cells carrying the gene. 1999 saw the start of a trial of intramuscular injection of the factor IX gene using an adeno-associated virus (AAV) vector.3It demonstrated that the gene had been successfully transferred, but factor IX levels did not increase. The authors concluded that the doses needed for success were too high to be delivered by intramuscular administration, and began work on developing administration into the liver. This proved successful, but the transplanted genes were destroyed by an immune response to the AAV vector.4
This apparently disappointing outcome was, in fact, a milestone. It showed that delivery of the virus into the liver was the route of administration of choice, and also that AAV was an effective vector. Treatment to suppress the immune response to the AAV vector was needed to preserve the gene, but it was soon found that oral prednisone was enough to do this – the immune response was transient and only a short course was needed. The principles for conducting future gene therapy trials had been established.
As of 2019, there are 13 clinical trials of gene therapy for haemophilia A or B under way, all using an adenovirus vector (though other viral vectors are also being evaluated). Three are Phase III trials designed to provide definitive evidence of efficacy and safety. Results so far show that factor VIII and IX activity after gene therapy is high and sustained, and that freedom from bleeding and its complications is now a realistic prospect.5,6
References
- Roth DA, Tawa NE Jr, O’Brien JM, Treco D, Selden RF; Factor VIII Transkaryotic Therapy Study Group. Nonviral transfer of the gene encoding coagulation factor VIII in patients with severe hemophilia A. N Engl J Med 2001; 344: 1735‒42.
- Doering CB, Spencer HT. Advancements in gene transfer-based therapy for hemophilia A. Expert Rev Hematol 2009; 2: 673‒83.
- Manno CS, Chew AJ, Hutchison S, et al. AAV-mediated factor IX gene transfer to skeletal muscle in patients with severe hemophilia B. Blood 2003; 101: 2963‒72.
- Manno CS, Pierce GF, Arruda VR, et al.Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat Med 2006; 12: 342‒7.
- Perrin GQ, Herzog RW, Markusic DM. Update on clinical gene therapy for hemophilia. Blood 2019; 133: 407‒14. doi: 10.1182/blood-2018-07-820720.
- Pierce GF, Coffin D; Members of the WFH Gene Therapy Round Table Program Committee and Organizing Committee. The 1st WFH Gene Therapy Round Table: Understanding the landscape and challenges of gene therapy for haemophilia around the world. Haemophilia 2019; 25: 189‒94. doi: 10.1111/hae.13673.
2000-


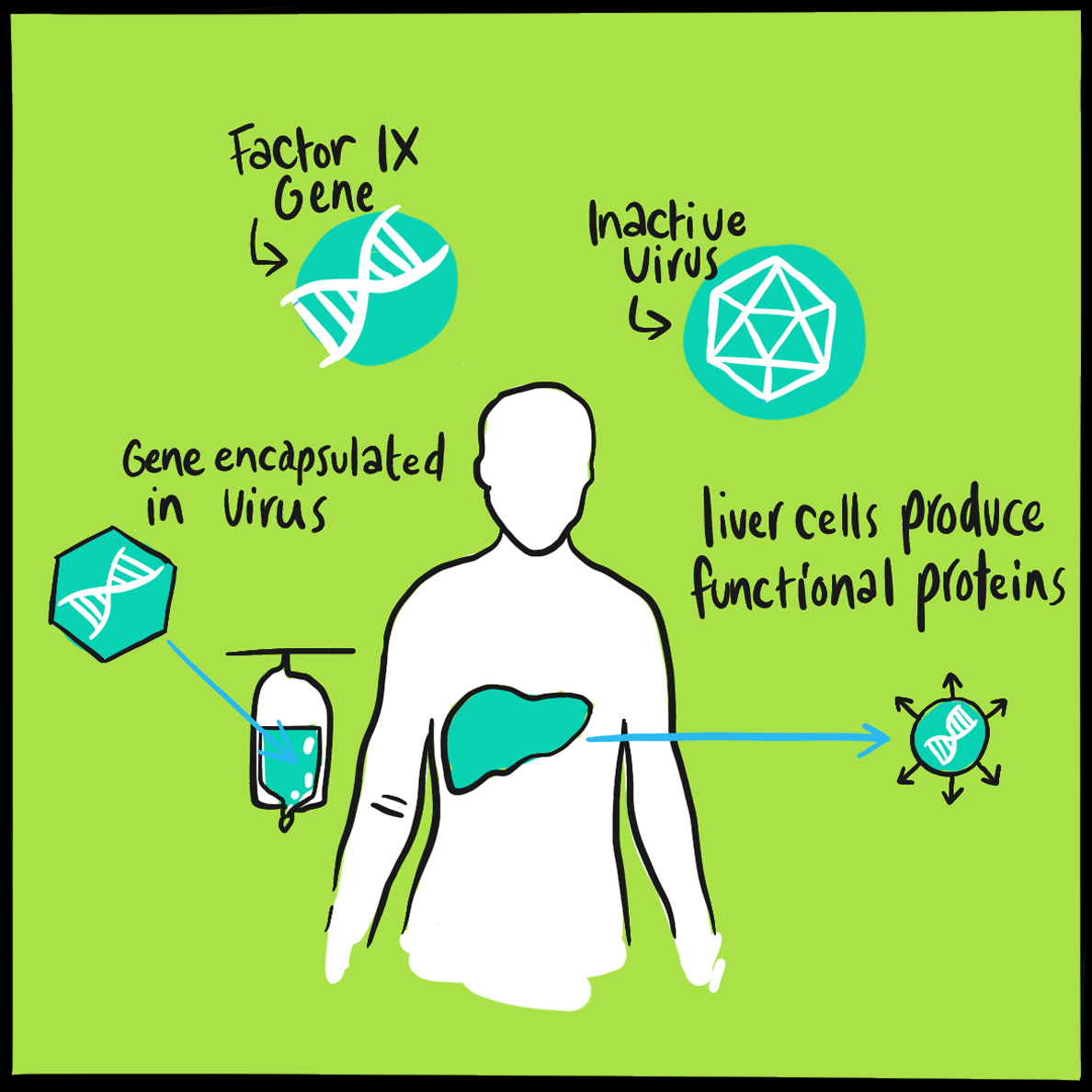

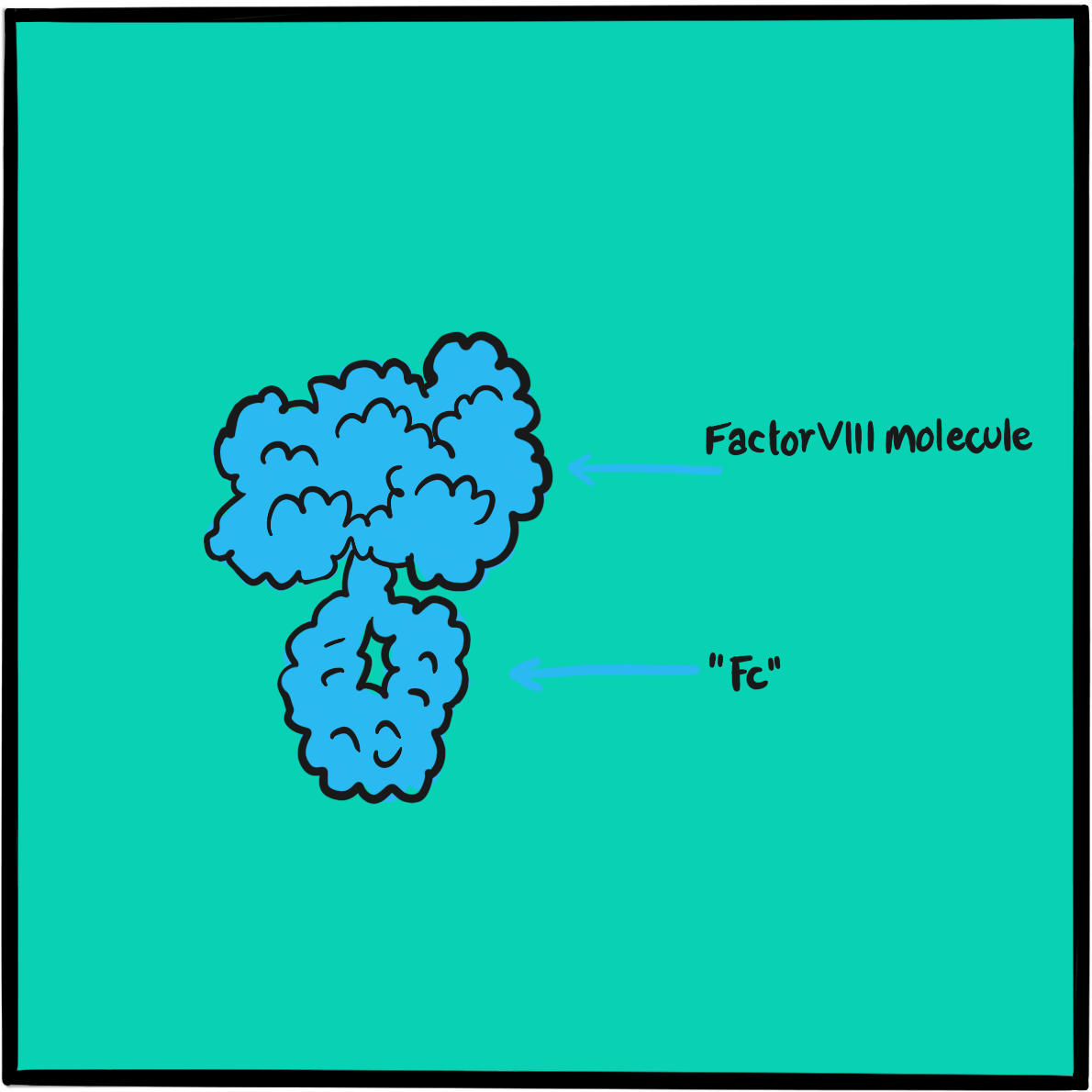
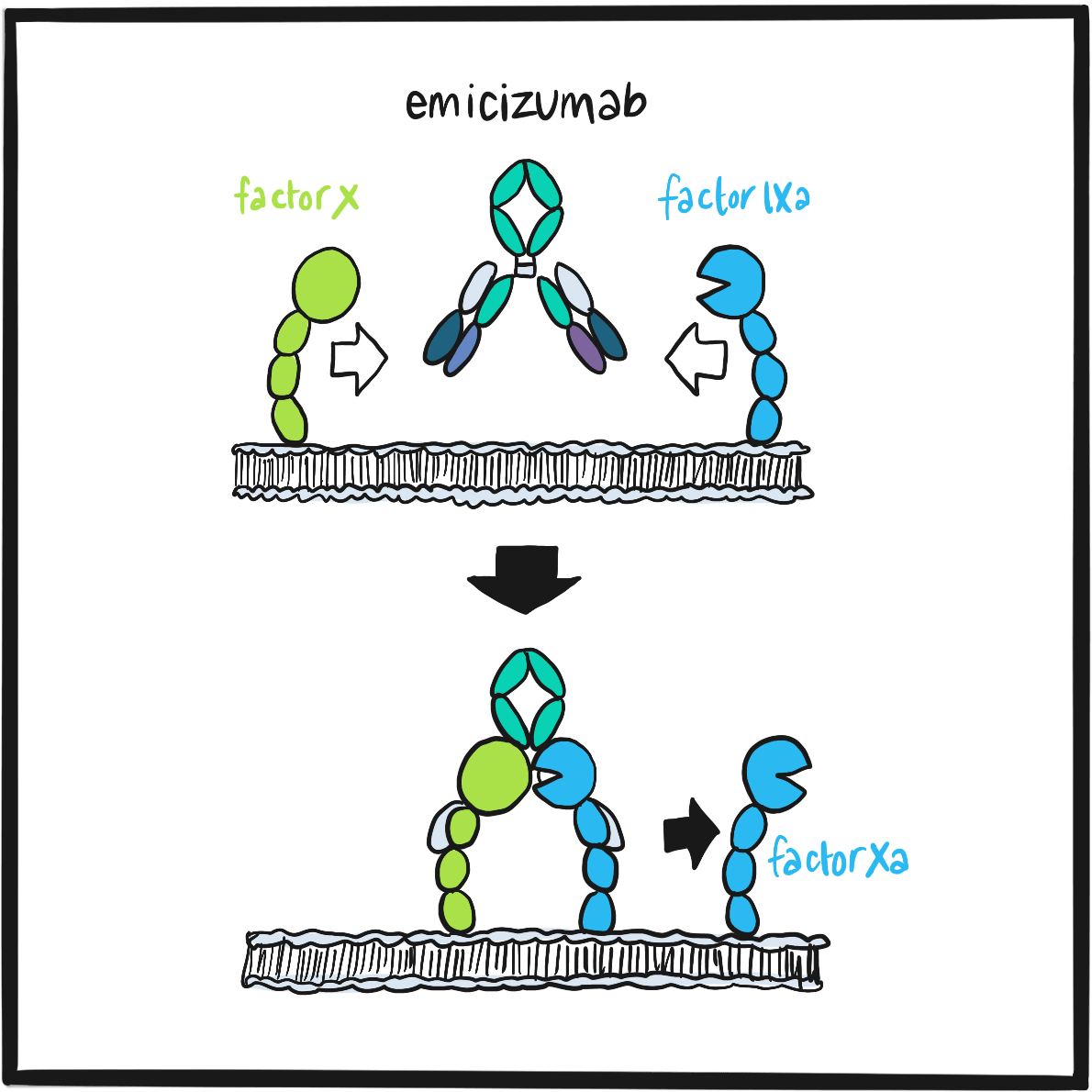
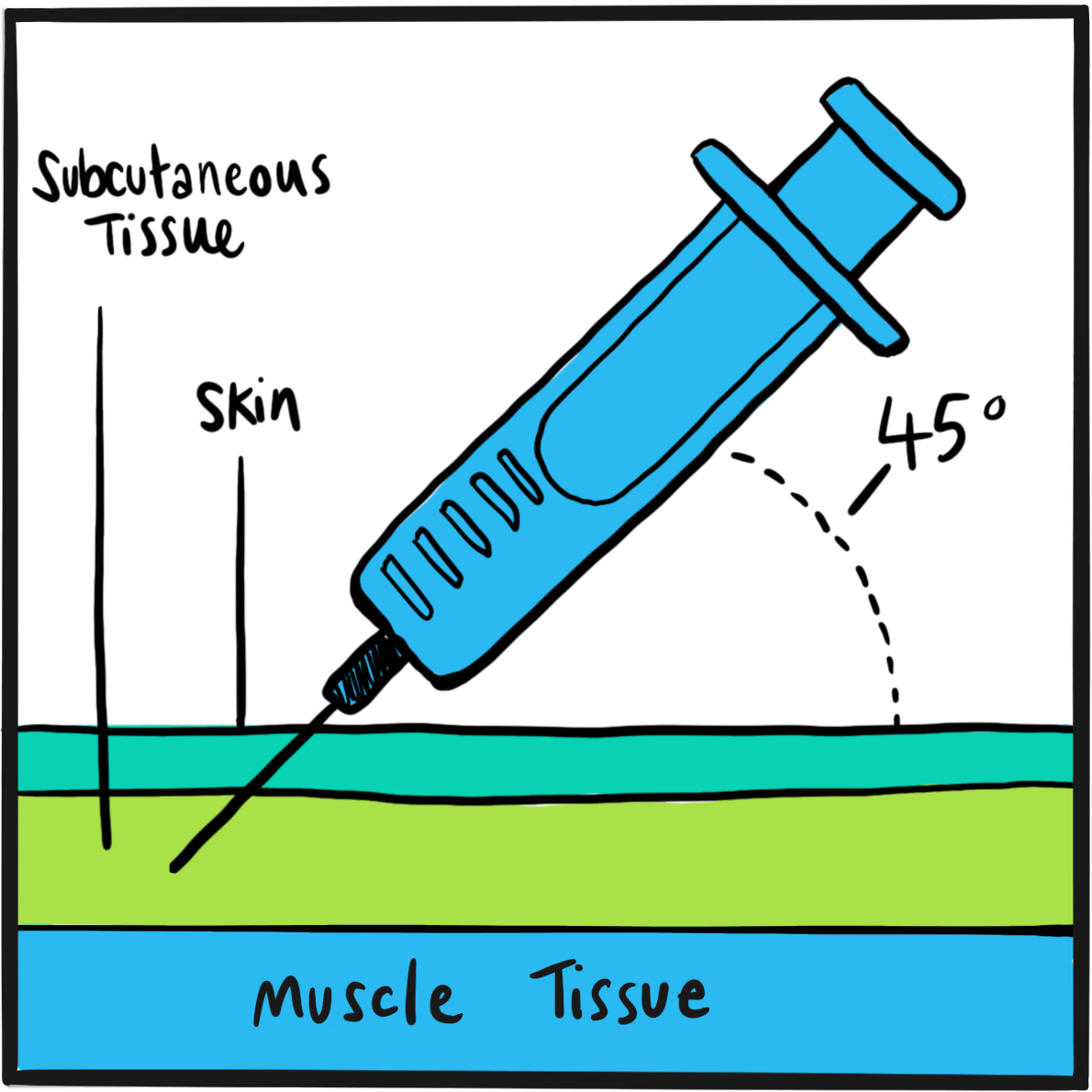
2007
The Joint Outcomes Study shows prophylaxis is effective read more
People with haemophilia who have repeated episodes of bleeding into joints develop disabling arthritis and experience constant pain ‒but for many years there was no way to prevent joint bleeds. Until the 1960s, factor replacement therapy required the infusion of large volumes of plasma and was feasible only for on-demand treatment of a bleed. The advent of factor concentrates allowed the use of much lower volumes. Small studies suggested that regular infusions (prophylaxis) could prevent bleeds, and that this might be a better strategy than waiting for a bleed to occur then treating it.
This promising approach was foiled by the contamination of blood products with HIV and hepatitis, and it wasn’t until the introduction of recombinant clotting factors (not derived from human blood and therefore free of viral contamination) that prophylaxis again became a serious option.
In 1991, a trial in Sweden showed that starting prophylaxis at age three was associated with less joint damage than starting at age five, and suggested that it would be even better to start at age one to two.1 Three years later, an American team showed that prophylaxis reduced the daily impact and complications of haemophilia by reducing joint bleeds.2
Definitive evidence of the benefits of prophylaxis, and of starting early in life, was first provided in 2007 by the Joint Outcome Study.3Sixty-five boys aged less than 30 months were randomly assigned to have standard on-demand treatment with recombinant factor VIII when a bleed occurred, or prophylaxis with infusions of recombinant factor VIII on alternate days. By six years of age, only 55% of boys treated on-demand still had normal joints, compared with 93% of those who received prophylaxis. The risk of joint damage detectable by an MRI scan was found to be six times greater in the on-demand group. There was a concern that regular exposure to factor VIII might increase the risk of inhibitors, and this occurred in two of the 32 boys using prophylaxis. On the other hand, three of the boys treated on-demand experienced a life-threatening haemorrhage during the study period.
Perhaps surprisingly, the Joint Outcome Study showed that joint damage did not correlate well with clinical signs of joint bleeding. The authors speculated ‘that chronic microhemorrhage into the joints or subchondral bone in young boys with hemophilia causes deterioration of joints without clinical evidence of hemarthroses and that prophylaxis prevents this subclinical process.’ And they were right. Prophylaxis from an early age ‒before the onset of joint damage, meaning before or soon after the first joint bleeds ‒is now the treatment of choice for haemophilia.4
References
- Petrini P, Lindvall N, Egberg N, Blombäck M. Prophylaxis with factor concentrates in preventing hemophilic arthropathy. Am J Pediatr Hematol Oncol 1991; 13: 280‒7.
- Aledort LM, Haschmeyer RH, Pettersson H. A longitudinal study of orthopaedic outcomes for severe factor-VIII-deficient haemophiliacs. The Orthopaedic Outcome Study Group. J Intern Med 1994; 236: 391‒9.
- Manco-Johnson MJ, Abshire TC, Shapiro AD, et al. Prophylaxis versus episodic treatment to prevent joint disease in boys with severe hemophilia. N Engl J Med 2007; 357: 535‒44. doi: 10.1056/NEJMoa067659.
- Ljung R, Gretenkort Andersson N. The current status of prophylactic replacement therapy in children and adults with haemophilia. Br J Haematol 2015; 169: 777‒86. doi: 10.1111/bjh.13365.
2008
Launch of Haemtrack, an electronic home treatment diary read more
Many young people and adults with a bleeding disorder now administer their treatment themselves at home or, if they are young children, their parents do it for them. Their medication and syringes arrive by courier, they reconstitute the factor and they give themselves the injection.
Most people prefer home treatment to visiting a hospital clinic several times a week. The downside is that there is less contact between the healthcare team and the person with the bleeding disorder and their family, so each treatment centre has procedures in place to ensure that patients have access to help and support when they need it, in addition to their regular clinic appointments. in the UK, week-by-week monitoring of treatment with clotting factor is managed via Haemtrack.
Haemtrack is an electronic diary accessed via a mobile phone app or website in which the patient (or parent) records details about the doses of clotting factor used, such as the day of administration and the batch number, and events such as bleeds. The app/website connects with the treatment centre and automatically transfers data so that it can be reviewed by the haemophilia nurse or doctor. Data can also be recorded on paper forms when the app/website are not suitable. Currently, about 90% of patients in the UK use Haemtrack.1
It is a condition of NHS home treatment that a patient’s treatment data are recorded using Haemtrack. Further, treatment with newer agents, such as extended half-life factors, is available only to individuals who adhere closely to the requirement for monitoring treatment with Haemtrack (that is, they record at least 75% of all infusions).
In addition to monitoring treatment, anonymised data from Haemtrack are used to support research about the use of clotting factors. For example, Haemtrack provided data for the THUNDER study, which revealed that people with moderate haemophilia may be undertreated with prophylactic clotting factor.2
References
- United Kingdom Haemophilia Centres Doctors’ Organisation and National Haemophilia Database. UKHCDO Annual report 2018 including bleeding disorder statistics for 2017/18. October 2018 (accessed April 2019).
- 2. Scott MJ, Xiang H, Hart DP, et al. Treatment regimens and outcomes in severe and moderate haemophilia A in the UK: The THUNDER study. Haemophilia 2019; 25: 205‒12. doi: 10.1111/hae.13616.
2011
Successful AAV vector-mediated gene transfer in haemophilia B read more
During the early 2000s, success with gene therapy for haemophilia was increasing incrementally rather than in leaps and bounds. By the middle of the decade, it was known that the genes should be targeted at the liver, encapsulated in a viral vector that could promote its uptake into the patient’s cells, from where it would start to produce clotting factor. The best viral vector was identified as adeno-associated virus (AAV) which, in its natural state, causes a benign infection in humans.
This, of course, raised the problem that individuals who had in the past had an AAV infection would have antibodies against the AAV vector and would not be eligible for gene therapy. There are several subtypes of AAV; they have some antigens in common, so that immunity against one may also confer protection against others. However, they are not equally prevalent in the population and the level of cross-sensitivity varies between subtypes.
The breakthrough trial of gene therapy for haemophilia B was published in 2011.1 A joint research team from the UK and the US developed an approach that they said was different from earlier attempts in three ways. First, they modified the vector to increase gene expression. Second, they used the AAV8 virus subtype, one of the less common naturally occurring subtypes in humans and therefore associated with a lower rate of background immunity. The AAV8 subtype also has an exceptionally strong affinity for liver cells, which enabled the third innovation of administering the gene/vector via a peripheral vein rather than directly into the liver.
The trial involved three pairs of people with severe haemophilia B (FIX <1%), who received low, medium and high doses. After 6–16 months, FIX activity levels were 2%‒11%. Four participants were able to discontinue factor IX prophylaxis completely and had no more bleeds; the other two were able to reduce their factor dose frequency. The two who received the highest dose had evidence of an immune response to the AAV8 vector; this was treated with a short course of steroids.
The trial was published on 22 December 2011 in one of the most prestigious medical journals, The New England Journal of Medicine, accompanied by a commentary wittily titled ‘Merry Christmas for haemophilia B patients’ (haemophilia B was originally known as Christmas disease, after the family name of the boy in whom it was first formally described). This commentary referred to the trial as ‘truly a landmark study’ and answered the question ‘Should the practicing hematologist rush to order this gene therapy vector if it is approved by the Food and Drug Administration?’ with the reply: ‘The answer is probably yes, but the risks of this procedure are not yet totally clear.’ This more or less sums up the state of knowledge several years later, in 2019.
Reference
- Nathwani AC, Tuddenham EG, Rangarajan S, et al. Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N Engl J Med 2011; 365: 2357‒65. doi: 10.1056/NEJMoa1108046.
2013
Haemnet is registered as a charity read
more
Haemnet supports a vibrant and interactive community of nurses, physiotherapists and allied healthcare professionals, sharing knowledge and experiences, and engaging in research that enables people affected by bleeding disorders to live well.
Haemnet began as an online professional network for the nurses and healthcare professionals who look after people with haemophilia and inherited bleeding disorders. Before Haemnet, when a haemophilia nurse needed advice or encountered a ‘difficult-to-manage’ patient, he or she would phone a friend for advice. Now, those conversations also take place on Haemnet with more voices contributing, to the benefit of the whole community. What began as a UK-wide initiative quickly spread across the global haemophilia community: now more than 550 nurses and physiotherapists from around the world are registered on Haemnet, along with a growing number of psychologists and social workers.
In 2013, in response to the views of members of the Haemophilia Nurses Association (HNA), Haemnet became a registered charity, overseen by a board of trustees, with the aim of providing education and collaborative research opportunities for all members of the haemophilia multidisciplinary care team. Day-to-day management of Haemnet is the responsibility of the director, Mike Holland, with strategic development support from Sandra Dodgson.
Haemnet runs the annual HNA conference alongside a growing number of haemophilia-specific and professional development courses. The courses are the result of listening to members in the context of the future of haemophilia care and identifying areas for development. They include the Contemporary Care of People with Bleeding Disorders course, a four-day residential training course for healthcare professionals, and ASPIRE, a leadership development programme followed over six to eight months.
The outputs and outcomes of Haemnet’s projects are shared with practitioners on the Haemnet website (which also provides CPD opportunities and a forum for HNA members), and through The Journal of Haemophilia Practice (JHP). Haemnet launched JHP as an open-access, online journal for haemophilia care professionals in 2014, and has now published over 100 articles. The journal is currently supported by charitable donations from Sobi, CSL Behring, Octapharma, Pfizer, Novo Nordisk and Grifols.
2015
First extended half -life factor VIII approved in Europe read more
Prophylaxis with clotting factor replacement therapy has been a tremendous leap forward for people with haemophilia: it greatly reduces bleeds and lowers the risk of joint damage compared with on-demand treatment. But it comes at the considerable cost of frequent infusions. As a result of pharmacokinetics ‒or the rate at which a substance is cleared from the body ‒most people with haemophilia A have to inject themselves with factor VIII on alternate days or two to three times per week, with some needing daily injections.1,2 This is time-consuming, and parents find it stressful having to administer infusions to their children.
Reducing the frequency of infusions would therefore be a major step forward ‒one that could make a big difference to a person who has to inject daily or every other day. Extended half-life (EHL) factors promise exactly that.
Half-life is a measure of how quickly factor is cleared from the body, measured as the time taken for its concentration in blood to fall by half. EHL factors are clotting factors that have been modified by adding side chains to the molecule, slowing the rate at which they are cleared from the body. This means that a dose lasts longer, prolonging the time until the next injections is needed.
On 19 November 2015, the first EHL factor VIII was approved for use in Europe. Efmoroctocog alfa, better known as Elocta, was developed in the United States by Biogen and marketed by Sobi. It is essentially a modified recombinant factor VIII (its B domain is removed, as in the conventional product ReFacto AF) that is fused to part of a protein (human immunoglobulin). Its half-life averages 19–21 hours in adults and 12–18 hours in children. This compares with 11–12 hours and 9 hours respectively for a conventional factor VIII. In clinical trials, efmoroctocog alfa was administered every three to five days and still maintained factor VIII activity above trough level for longer than a conventional product. In February 2018, the Republic of Ireland announced that all people with haemophilia would be switched from a conventional factor VIII to an EHL product.
To date, clinical trials have compared EHL factors with historical experience with conventional factors. These data suggest they reduce the frequency of bleeds with less frequent injections. However, the question for each individual switching from a conventional to an EHL factor is whether to aim for the same trough factor VIII activity level and have fewer injections, or to have a higher (and safer) trough level and keep injection frequency the same.
As of April 2019, one other EHL factor VIII product had been licensed in Europe: rurioctocog alfa pegol (Adynovi) is a factor VIII combined with a polyethylene glycol (PEG) side chain. Novo Nordisk’s turoctocog alfa pegol (Esperoct), which combines factor VIII with PEG, has been licensed in the United States.
Three EHL factor IX products are available: eftrenonacog alfa (factor IX fused to immunoglobulin; Alprolix), nonacog beta pegol (factor IX combined with PEG; Refixia); and albutrepenonacog alfa (factor IX fused to albumin; Idelvion).
References
- Björkman S, Oh M, Spotts G, et al. Population pharmacokinetics of recombinant factor VIII: the relationships of pharmacokinetics to age and body weight. Blood 2012; 119: 612‒8. doi: 10.1182/blood-2011-07-360594.
- Berntorp E, Dolan G, Hay C, et al. European retrospective study of real-life haemophilia treatment. Haemophilia 2017; 23: 105‒14. doi: 10.1111/hae.13111.
Further reading
Graf L. Extended half-life factor VIII and factor IX preparations. Transfus Med Hemother 2018; 45: 86‒91. doi: 10.1159/000488060.
Collins P, Chalmers E, Chowdary P, et al. The use of enhanced half-life coagulation factor concentrates in routine clinical practice: guidance from UKHCDO. Haemophilia 2016; 22: 487‒98. doi: 10.1111/hae.13013.
2017
Emicizumab, a humanized bispecific antibody, approved by the FDA read more
Clotting factors are large proteins. When they are infused, there is a risk that they will provoke an immune reaction that will lead to the formation of antibodies, which bind to the clotting factor, neutralising its activity. In the management of haemophilia, neutralising antibodies are known as inhibitors.
Inhibitors are more frequently a problem for people with haemophilia A than those with haemophilia B as the factor VIII molecule is larger and more immunogenic than factor IX. Inhibitors may occur at a low or high level. Low-level inhibitors can be overcome by increasing the dose of clotting factor; high-level inhibitors render a clotting factor useless. A person who develops high-level inhibitors needs additional treatment with a preparation that circumvents the need for factor VIII or IX. This may be factor VII or a preparation of mixed factors known as FEIBA (factor eight bypassing activity). Alternatively, they can undergo immune tolerance induction with very high doses of clotting factor designed to overwhelm their immune response. However, this can take months of daily infusions and is very expensive. Emicizumab has changed all that.
Emicizumab is a monoclonal antibody that takes the place of factor VIII in the coagulation cascade by acting as a bridge between factor IX and factor X. It does not resemble factor VIII in structure and does not induce neutralising antibodies. It makes factor VIII redundant and inhibitors irrelevant.
In 2017, emicizumab (Hemlibra, Roche) was approved by the US medicines regulator, the Food and Drug Administration (FDA), for routine prophylaxis in children and adults with haemophilia A with inhibitors. It was approved for use in Europe on 23 February 2018.
Further reading
Barg AA, Livnat T, Kenet G. Inhibitors in hemophilia: treatment challenges and novel options. Semin Thromb Hemost 2018; 44: 544‒50. doi: 10.1055/s-0037-1612626.
Rodriguez-Merchan EC, Valentino LA. Emicizumab: review of the literature and critical appraisal. Haemophilia 2019; 25: 11‒20. doi: 10.1111/hae.13641.
2018
Subcutaneous therapy available for people with inhibitors in UK read more
On 23 February 2018, emicizumab was approved by the European Medicines Agency as routine prophylaxis for bleeding episodes in children and adults with haemophilia A with factor VIII inhibitors. It is an alternative to the burdensome and expensive approaches to managing inhibitors previously available: treatment with a bypassing agent (factor VII or FEIBA (factor eight bypassing activity)) or immune tolerance induction, an intensive regimen of daily infusions of high-dose clotting factor designed to overwhelm the immune response.
Treatment with emicizumab is an altogether different prospect. It is administered by subcutaneous injection, initially weekly; but after a month this can be reduced to every two weeks or monthly. All other treatment with a clotting factor can be stopped. Furthermore, emicizumab proved more effective than bypassing agents at preventing bleeds: in the key clinical trial providing the evidence for its authorisation, weekly emicizumab was associated with a 79% lower bleeding rate than had been achieved through prophylaxis with bypassing agents.1
In 2018, emicizumab was also approved as prophylaxis in people with severe haemophilia A who did not have inhibitors, thereby replacing the need for 90–365 intravenous infusions every year with only 12–52 subcutaneous injections.
In July 2018, NHS England announced that every patient with haemophilia A with inhibitors and poorly controlled bleeding would be eligible for treatment with emicizumab. On 11 March 2019, the European Medicines Agency approved emicizumab for the treatment of people with severe haemophilia A whether they had inhibitors or not.
In an era where new technologies promise radical change, emicizumab is a truly disruptive innovation. Real-world experience of its use is currently limited; however, if the evidence of efficacy and safety is borne out in day-to-day clinical practice, it could transform care for people with haemophilia A. Its use does not need the close support that is customary with clotting factor replacement, which has implications not only for patients but also for haemophilia treatment centres. Emicizumab’s future depends on how cost is balanced against effectiveness and potentially improved quality of life, compared with the familiar options that have served the haemophilia community well for the previous 20 years.
Reference
- Oldenburg J, Mahlangu JN, Kim B, et al. Emicizumab prophylaxis in hemophilia A with inhibitors. N Engl J Med 2017; 377: 809‒18. doi: 10.1056/NEJMoa1703068.
Further reading
Barg AA, Livnat T, Kenet G. Inhibitors in hemophilia: treatment challenges and novel options. Semin Thromb Hemost 2018; 44: 544‒50. doi: 10.1055/s-0037-1612626.